
R/Bioconductor를 활용한 Pathway 맵 그리기
- Posted at 2015/04/03 18:02
- Filed under 생물정보
RNA-seq(발현분석) 후 관심 있는 유전자 발현에 따른 증가/감소한 유전자들이 어떤 Pathway에 속하는지 알아보고자 할
경우 Pathway 맵핑을 통하여 확인할 수 있다. Pathway 맵핑과 시각화를 지원하는 무료 프로그램은 KEGG Mapper 와 같은 웹 어플리케이션과 Blast2GO , Cytoscape 같은 맵핑 및 네트워크 분석 프로그램이 알려져 있다. 그 중에 Bioconductor에서 패키지로 제공하는 R로 작성된 Pathview 도구의 설치 및 활용법에 대해 알아보겠다.
Pathway map 그리기 초급편
설치가 다 되었다면 간단한 소스코드를 이용하여, 맵정보를 그려보자.
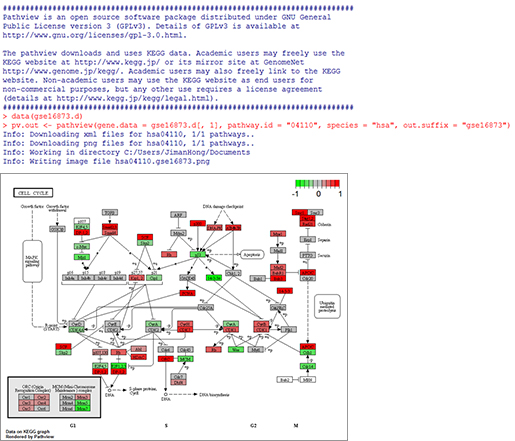
위 코드를 실행해보면 아래 <그림2>와 같이 Pathview 라이브러리를 불러오고, KEGG를 통하여 맵정보를 다운받은 후 테스트 데이터의 발현정보를 Pathway(KEGG04110 - Cell cycle - Homo sapiens) 맵정보에 반영하여 새로운 맵정보를 만들어 파일로 출력한다.
데이터를 만든 후 아래의 소스코드로 R 커맨드 창에서 실행해보자.
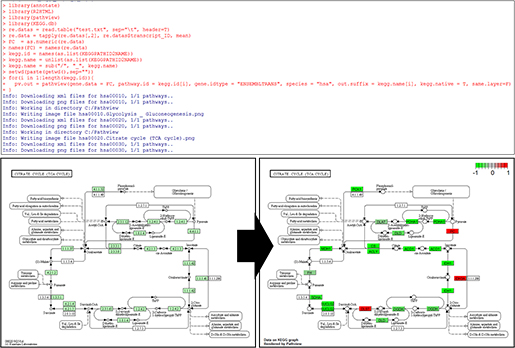
아래 그림4와 같이 소스코드를 실행하면, 입력한 발현정보에 대한 Human 종 전체를 대상으로 한 새로운 Pathway맵이 만들어지고 자동으로 이미지로 출력된다.
<그림1> Pathview Bioconductor 메인페이지
Pathview는 KEGG 맵의 원하는 유전자 위치를 직관적으로 확인할 수 있도록 지원하는
시각화 프로그램으로 발현분석 후 특정 유전자의 Pathway를 확인하는데 유용한 프로그램이다. Pathview와 기존에 알려진
다른 시각화 프로그램과의 다른점은 발현정보의 반영이다. 기존의 시각화 프로그램은 Pathway 상에서 원하는 유전자의 위치 및
군집 등의 정보를 확인 할 수 있는 반면에 Pathview를 이용하면, Pathway 맵을 통하여 실제 발현이 증가하였는지
감소하였는지를 확인할 수 있고, 그 발현정도의 차이도 확인이 가능하다. 다만 R 소스 기반이라서 프로그램 언어에 대한 장벽이
있으나, 이번 시간에는 예제를 통해 비교적 간단히 사용할 수 있는 활용법을 정리해 본다.
설치시 필요한 라이브러리
R/Bioconductor 팩키지이므로 사용하기 위해서는 R을 설치하여야 한다. 최신 버젼은 설치시 기존에 사용하는 라이브러리와 충돌이 발생할 수 있기 때문에 버젼에 맞게 설치해야 한다. 이번 테스트는 R3.1.1을 기준으로 진행하였다. 또한 pathview 를 포함한 추가적인 라이브러리가 필요하기 때문에, 총 4개의 라이브러리를 설치 후 진행해보도록 한다.
설치시 필요한 라이브러리
R/Bioconductor 팩키지이므로 사용하기 위해서는 R을 설치하여야 한다. 최신 버젼은 설치시 기존에 사용하는 라이브러리와 충돌이 발생할 수 있기 때문에 버젼에 맞게 설치해야 한다. 이번 테스트는 R3.1.1을 기준으로 진행하였다. 또한 pathview 를 포함한 추가적인 라이브러리가 필요하기 때문에, 총 4개의 라이브러리를 설치 후 진행해보도록 한다.
줄 번호 보이기/숨기기
1 #library 'annotate' 설치
2 >source("http://bioconductor.org/biocLite.R")
3 >biocLite("annotate")
4
5 #library 'R2HTML' 설치 - 통합 package 'ArrayTool'로 대체(또는
R 내부 라이브러리 이용)
6 >source("http://bioconductor.org/biocLite.R")
7 >biocLite("ArrayTools")
8
9 #library 'pathview' 설치
10 >source("http://bioconductor.org/biocLite.R")
11 >biocLite("pathview")
12
13 #library 'KEGG.db' 설치
14 >source("http://bioconductor.org/biocLite.R")
15 >biocLite("KEGG.db")
Pathway map 그리기 초급편
설치가 다 되었다면 간단한 소스코드를 이용하여, 맵정보를 그려보자.
줄 번호 보이기/숨기기
1 library(pathview)
2 data(gse16873.d)
3 pv.out <- pathview(gene.data = gse16873.d[, 1], pathway.id =
"04110", species = "hsa", out.suffix = "gse16873")
위 코드를 실행해보면 아래 <그림2>와 같이 Pathview 라이브러리를 불러오고, KEGG를 통하여 맵정보를 다운받은 후 테스트 데이터의 발현정보를 Pathway(KEGG04110 - Cell cycle - Homo sapiens) 맵정보에 반영하여 새로운 맵정보를 만들어 파일로 출력한다.
<그림2> Pathview 실행 및 맵 그리기
Pathway map 그리기 응용편
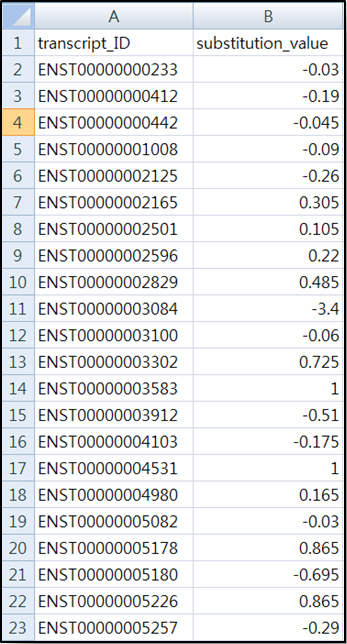
이번에는 직접 데이터를 만든 후 입력해보자, 데이터는 그림3과 같이 엑셀형태로 만들 수 있다.
이번에는 직접 데이터를 만든 후 입력해보자, 데이터는 그림3과 같이 엑셀형태로 만들 수 있다.
<그림3> 발현정보를 반영한 입력데이터 만들기
데이터를 만든 후 아래의 소스코드로 R 커맨드 창에서 실행해보자.
줄 번호 보이기/숨기기
1 library(annotate)
2 library(R2HTML)
3 library(pathview)
4 library(KEGG.db)
5 re.datas = read.table("test.txt", sep="\t", header=T)
6 re.data = tapply(re.datas[,2], re.datas$transcript_ID, mean)
7 FC = as.numeric(re.data)
8 names(FC) = names(re.data)
9 kegg.id = names(as.list(KEGGPATHID2NAME))
10 kegg.name = unlist(as.list(KEGGPATHID2NAME))
11 kegg.name = sub("/", "_", kegg.name)
12 setwd(paste(getwd(),sep=""))
13 for(i in 1:length(kegg.id)){
14 pv.out = pathview(gene.data = FC, pathway.id = kegg.id[i], gene.idtype = "ENSEMBLTRANS", species = "hsa", out.suffix = kegg.name[i], kegg.native = T, same.layer=F)
15 }
아래 그림4와 같이 소스코드를 실행하면, 입력한 발현정보에 대한 Human 종 전체를 대상으로 한 새로운 Pathway맵이 만들어지고 자동으로 이미지로 출력된다.
<그림4> Human 종을 대상으로 Pathway 맵 시각화 진행
Pathview - KEGG map 연결정보
Pathview는 KEGG 내의 맵정보를 이용하기 때문에 KEGG에서 제공하지 않는 종들에게는 적용이 쉽지 않지만, 모듈에서 제공하는 19개의 모델종에 대해서는 적용이 잘된다. 또한 지원가능한 ID 형태가 제한적이기 때문에 사용시 ID를 적절히 변환하여야 한다.
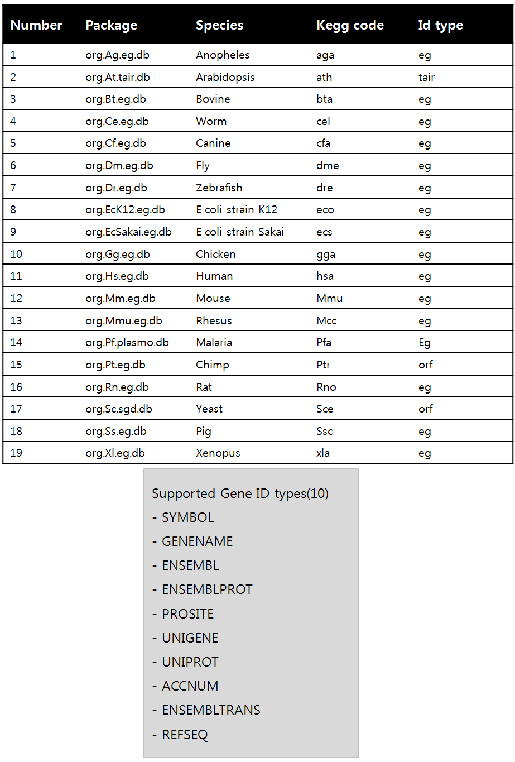
Pathview는 KEGG 내의 맵정보를 이용하기 때문에 KEGG에서 제공하지 않는 종들에게는 적용이 쉽지 않지만, 모듈에서 제공하는 19개의 모델종에 대해서는 적용이 잘된다. 또한 지원가능한 ID 형태가 제한적이기 때문에 사용시 ID를 적절히 변환하여야 한다.
<그림5> pathview에서 활용가능한 모델종 정보 및 ID 타입
Pathview를 잘 활용할 수 있는 노하우
Pathview를 활용한 pathway 맵 그리기는 19개의 모델종으로 제한적이기에 NGS 기반의 de novo transcripts 등에는 적절하지 않다. 그러나 상동성기반으로 ID를 맵핑하거나 변환하면, 19개 모델종의 근연종에도 적용이 가능하기에 원하는 특정 기작연구에 가이드로 활용될 수 있을 것이다.
Pathview를 활용한 pathway 맵 그리기는 19개의 모델종으로 제한적이기에 NGS 기반의 de novo transcripts 등에는 적절하지 않다. 그러나 상동성기반으로 ID를 맵핑하거나 변환하면, 19개 모델종의 근연종에도 적용이 가능하기에 원하는 특정 기작연구에 가이드로 활용될 수 있을 것이다.
작성자 : 데이터사이언스센터 통합개발실
주임 개발자 홍지만
주임 개발자 홍지만
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/176
Trackback URL : 이 글에는 트랙백을 보낼 수 없습니다

