첫 직장, 그리고 한달
- Posted at 2010/02/12 17:04
- Filed under 회사소식
우리회사 신입사원의 이야기입니다. "훈남"으로 통하면서 여직원들의 관심을 독차지하고 있다죠. (질투 삐릿~) 입사 후 한달이 지난 그의 이야기를 들어볼까 합니다.
KM팀 신입사원 이재영
인실리코젠에 지원서를 제출하고 약 1주간의 기다림 끝에 면접을 보게 되었습니다. 대학에서의 마지막 시험 기간과 일정이 겹쳐져 학업과 면접을 동시에 준비하는 상태가 되어 이 한 주 동안에만 살이 2~3kg정도 빠졌던 기억이 납니다.
드디어 면접 당일, 이른 아침에 일어나 준비하던 기분이 아직도 생생합니다. 긴장과 기대, 두려움과 설렘 같은 기분들이 복잡스럽게 뒤엉키며 저의 마음을 흔들었고 혹여 늦지 않을까 하는 걱정에 이른 걸음을 하다보니 면접시간보다 1시간 정도 먼저 도착했습니다. 잠시 동안의 주변을 둘러 본 이후 회사의 문을 두드렸을 때 일찍 오셨다는 말과 함께 문을 열어주셨던 분이 경윤 주임님 이셨죠. 그리고 곧 면접, 김 팀장님과 박 팀장님 두 분이 들어오셨습니다. 두 분과의 긴 면접을 마치고 집으로 돌아오는 길은 아쉬움과 홀가분함이 반반인 기분의 짧은 길이였습니다.
합격통지를 받고 너무 큰 설렘에 답을 보내는 것을 잊어 임 실장님께서 따로 전화를 하시는 수고를 만들어 드리기도 했습니다. 이후 첫 출근에서의 어색함과 미숙함은 다양한 실수들을 만들기도 했고 그러는 가운데 본격적인 업무가 시작되었습니다.
처음으로 팀에 들어와서 진행된 프로젝트는 정행사 논문투고 시스템(OJMS)입니다. 웹 방면의 코딩은 부족한 상태이기에 코드의 구현 보다는 자료의 조사나 정리, 요약과 같은 서브 작업들을 위주로 진행 중입니다. 현재는 화면 구성을 하고 있습니다.
 OJT 기간 중 처음으로 만져본 코드인 혈연관계정보검색시스템(KinMatch)입니다. 맨땅에 헤딩하는 기분으로 약간의 기본만을 가진 상태로 모든 파일을 열어보며 구조를 확인하고 그것들을 하나하나 바꾸어 가며 각각의 경우에 변하는 화면을 보면서 수정해 나갔습니다. 이로 인해 장고(Django)를 이용해 만드는 웹페이지의 구조와 프로젝트 진행 절차 및 subversion의 사용법, vi editor의 숙달 그리고 CSS와 HTML의 기본을 익혔습니다. 그리고 혼자 진행하는 것이 아닌 Descign팀과의 협업으로 진행해 나가 업무협조의 과정과 방법을 익히게 해준 과정입니다. 그리고 단순히 여기서 종료되지 않고 한국저작권 위원회에 프로그램 등록을 통해 지적 재산권의 확보와 사업의 진행을 위한 기본적인 과정을 익혔습니다.
OJT 기간 중 처음으로 만져본 코드인 혈연관계정보검색시스템(KinMatch)입니다. 맨땅에 헤딩하는 기분으로 약간의 기본만을 가진 상태로 모든 파일을 열어보며 구조를 확인하고 그것들을 하나하나 바꾸어 가며 각각의 경우에 변하는 화면을 보면서 수정해 나갔습니다. 이로 인해 장고(Django)를 이용해 만드는 웹페이지의 구조와 프로젝트 진행 절차 및 subversion의 사용법, vi editor의 숙달 그리고 CSS와 HTML의 기본을 익혔습니다. 그리고 혼자 진행하는 것이 아닌 Descign팀과의 협업으로 진행해 나가 업무협조의 과정과 방법을 익히게 해준 과정입니다. 그리고 단순히 여기서 종료되지 않고 한국저작권 위원회에 프로그램 등록을 통해 지적 재산권의 확보와 사업의 진행을 위한 기본적인 과정을 익혔습니다.
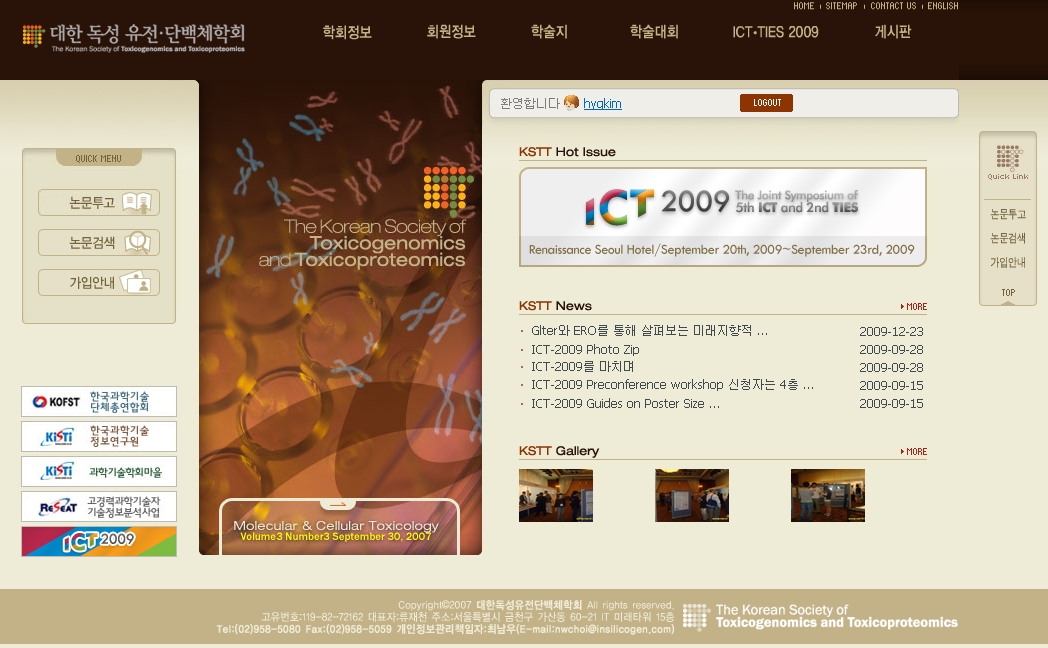 마지막으로 현재 기획중인 대한독성유전단백체학회의 홈페이지 리뉴얼 작업입니다. 이 부분에서는 단지 프로그램만을 제작하는 과정만이 아닌 기획서의 작성을 통해 프로젝트 진행 이전 단계를 알아가고 있습니다. 또한 기획을 하며 사람들이 필요로 하는 기능이 무엇이 있는지를 생각하는 과정을 거침으로 해서 내가 앞으로 알아야 할 부분이 어떤 것인지, 그리고 그것을 구현하고 서비스 하는데 얼마만큼의 시간과 인력이 필요할지에 대해서도 생각해보게 되었습니다.
마지막으로 현재 기획중인 대한독성유전단백체학회의 홈페이지 리뉴얼 작업입니다. 이 부분에서는 단지 프로그램만을 제작하는 과정만이 아닌 기획서의 작성을 통해 프로젝트 진행 이전 단계를 알아가고 있습니다. 또한 기획을 하며 사람들이 필요로 하는 기능이 무엇이 있는지를 생각하는 과정을 거침으로 해서 내가 앞으로 알아야 할 부분이 어떤 것인지, 그리고 그것을 구현하고 서비스 하는데 얼마만큼의 시간과 인력이 필요할지에 대해서도 생각해보게 되었습니다.
한 달이라는 기간은 업무를 파악하는데 있어서 짧지 않은 기간입니다. 이 시간동안 회의도 많았고 다양한 주제의 대화도 오고 갔습니다. 이 가운데 제가 배워야 하는 것은 수 없이 많았지만 이들 중 얼마만큼을 찾았는가를 정확히 말씀드릴 수는 없습니다. 그러나 한 가지 확실한 점은 배워야 할 것이 많고 스스로 고쳐가야 할 것들 역시 많다는 것입니다. 그리하여 우선적으로 HTML, CSS, Django를 빠르게 익혀 실무에서 이용할 수 있도록 하는 것을 목표로 세우고 현재는 HTML에 집중하고 있습니다. 우선적으로 빠른 시간 안으로 HTML, CSS의 개괄적인 마무리를 하고 곧 Django의 정확한 개념 정립과 활용을 할 수 있을 정도의 실력을 갖추는 것을 목표로 세웠습니다. 이와 같은 개인적으로 부족한 부분을 보완하는 규칙적이며 꾸준한 과정과 동시에 회사에서 바로 필요한 것을 바로 얻을 수 있도록 빠르게 파악하며 익히는 단계를 진행시키면서 단시간 안에 열심히 하면서도 잘 하는 모습을 보여드리도록 하겠습니다.
KM팀 신입사원 이재영
인실리코젠에 지원서를 제출하고 약 1주간의 기다림 끝에 면접을 보게 되었습니다. 대학에서의 마지막 시험 기간과 일정이 겹쳐져 학업과 면접을 동시에 준비하는 상태가 되어 이 한 주 동안에만 살이 2~3kg정도 빠졌던 기억이 납니다.
드디어 면접 당일, 이른 아침에 일어나 준비하던 기분이 아직도 생생합니다. 긴장과 기대, 두려움과 설렘 같은 기분들이 복잡스럽게 뒤엉키며 저의 마음을 흔들었고 혹여 늦지 않을까 하는 걱정에 이른 걸음을 하다보니 면접시간보다 1시간 정도 먼저 도착했습니다. 잠시 동안의 주변을 둘러 본 이후 회사의 문을 두드렸을 때 일찍 오셨다는 말과 함께 문을 열어주셨던 분이 경윤 주임님 이셨죠. 그리고 곧 면접, 김 팀장님과 박 팀장님 두 분이 들어오셨습니다. 두 분과의 긴 면접을 마치고 집으로 돌아오는 길은 아쉬움과 홀가분함이 반반인 기분의 짧은 길이였습니다.
합격통지를 받고 너무 큰 설렘에 답을 보내는 것을 잊어 임 실장님께서 따로 전화를 하시는 수고를 만들어 드리기도 했습니다. 이후 첫 출근에서의 어색함과 미숙함은 다양한 실수들을 만들기도 했고 그러는 가운데 본격적인 업무가 시작되었습니다.
처음으로 팀에 들어와서 진행된 프로젝트는 정행사 논문투고 시스템(OJMS)입니다. 웹 방면의 코딩은 부족한 상태이기에 코드의 구현 보다는 자료의 조사나 정리, 요약과 같은 서브 작업들을 위주로 진행 중입니다. 현재는 화면 구성을 하고 있습니다.
한 달이라는 기간은 업무를 파악하는데 있어서 짧지 않은 기간입니다. 이 시간동안 회의도 많았고 다양한 주제의 대화도 오고 갔습니다. 이 가운데 제가 배워야 하는 것은 수 없이 많았지만 이들 중 얼마만큼을 찾았는가를 정확히 말씀드릴 수는 없습니다. 그러나 한 가지 확실한 점은 배워야 할 것이 많고 스스로 고쳐가야 할 것들 역시 많다는 것입니다. 그리하여 우선적으로 HTML, CSS, Django를 빠르게 익혀 실무에서 이용할 수 있도록 하는 것을 목표로 세우고 현재는 HTML에 집중하고 있습니다. 우선적으로 빠른 시간 안으로 HTML, CSS의 개괄적인 마무리를 하고 곧 Django의 정확한 개념 정립과 활용을 할 수 있을 정도의 실력을 갖추는 것을 목표로 세웠습니다. 이와 같은 개인적으로 부족한 부분을 보완하는 규칙적이며 꾸준한 과정과 동시에 회사에서 바로 필요한 것을 바로 얻을 수 있도록 빠르게 파악하며 익히는 단계를 진행시키면서 단시간 안에 열심히 하면서도 잘 하는 모습을 보여드리도록 하겠습니다.
Posted by 人Co
- Tag
- 신입사원
- Response
- No Trackback , 1 Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/38





 입사지원서.doc
입사지원서.doc