CLC Genomics Workbench V.23 Release Note
New tools
Homology Based Cloning
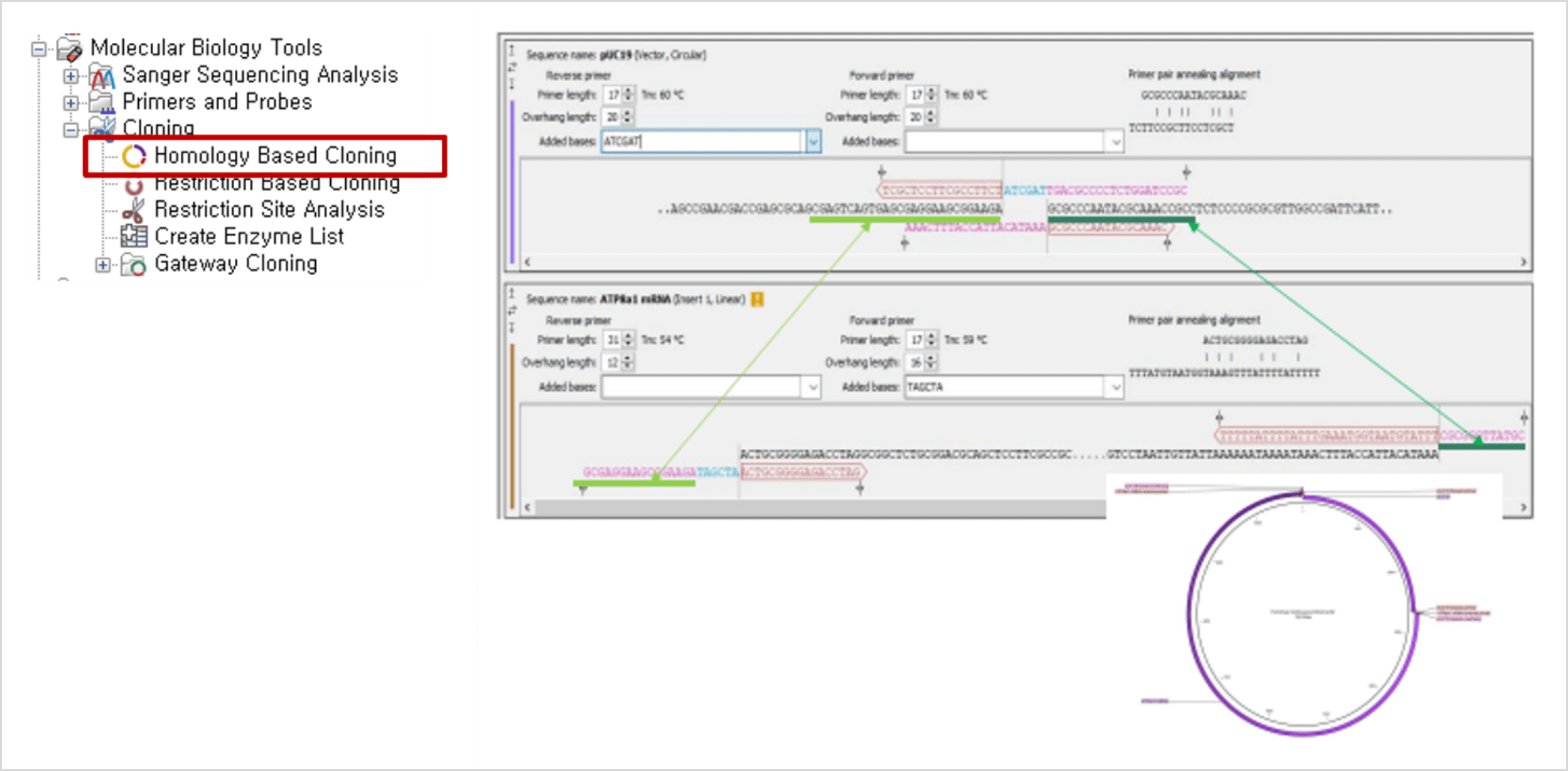

- Gibson Assembly®와 같은 homologous ends에 의존하는 Cloning 방법을 위한 Cloning 실험을 설계합니다.
- genes/transcripts/miRNA 등의 특징 클러스터를 찾습니다. Sankey 플롯, 그래프 등 시각화된 결과를 함께 포함합니다.
New tools coming from plugins
Detect and Refine Fusion Genes
- 잠재적인 fusion을 식별한 다음 정제하여 RNA-Seq 데이터에서 fusion genes를 찾습니다.
- 여러 샘플의 Coverage를 분석하고 비교합니다.
- Variant Track 및 Reference Sequence에서 Consensus Sequences를 생성합니다.
- GFF, GVF 또는 GTF 형식의 파일을 Sequences List에 Annotation합니다. 이 Tool는 이전에 Annotate with GFF file Plug in 으로 배포되었습니다.
Other new functionality and improvements
RNA-Seq analysis tools
RNA-Seq Analysis
- Transcript Expression(TE) Table에서 유전자 명(중복 제외)이 변경되지 않습니다. 이전에는 유전자 명의 숫자를 더한 이름(예: “BRCA_1”)으로 변경되었습니다. 따라서, CLC Genomics Workbench 23버전의 TE Track을 히트 맵, PCA 플롯, 표현식 등을 생성함에 있어 이전 버전의 TE Track과 함께 사용할 수 없습니다.
- Input Reads가 Biomedical Genomics Analysis Plug in에 의해 UMI로 annotation 되고, Library 유형이 RNA-Seq 분석을 위한 3’ Sequencing으로 설정된 경우 보고서의 Fragment statistics section에 UMI의 수가 포함됩니다.
- 샘플들은 Tree, 샘플 또는 활성 metadata layer options 이나 개별 Metadata 항목별로 정렬할 수 있습니다.
- Features를 재정렬 하여 왼쪽 위에서 오른쪽 아래로 대각선을 만드는 새로운 옵션입니다.
- Metadata 카테고리의 순서를 조정할 수 있습니다.
- 유전자를 시각화하기 위한 새로운 Plot이 포함되어 있으며, Metadata 카테고리 등은 알파벳순으로 정렬됩니다.
- 4개 및 5개의 그룹의 분석을 지원 합니다. (이전까지는 최대 3개 그룹)
- 두 개의 Table이 Output 되며, 첫 번째 Table에는 PCA 데이터 View가 나타나고, 두 번째 Table에는 점의 좌표로 표시 됩니다.
- 2D PCA Plot 의 Metadata 카테고리의 순서를 조정할 수 있습니다.
miRNA analysis tools
Quantify miRNA
- 중복된 이름을 포함하는 데이터베이스(Customize)를 가공합니다.
- 60bp보다 긴 시퀀스를 포함하는 데이터베이스(Customize)를 허용하지 않습니다. (SmallRNAs 와 유사한 시퀀스에 대한 Reads의 잘못된 할당을 방지할 수 있습니다.)
- 여러 inputs를 추가할 때 Expression Tables에는 샘플 중에서 식별된 “Combined set of IsomiRs” 에 대한 항목이 포함되어 Differential Expression in Two Groups 및 Differential Expression for RNA-Seq 분석에 호환됩니다.
Differential Expression for RNA-Seq and Differential Expression in Two Groups
- Differential Expression for RNA-Seq 및 Differential Expression in Two Groups에서 생성된 miRNA Statistical Comparison Table에 subset을 위한 새로운 옵션이 추가 되었습니다.
- Outliers를 낮출 수 있습니다. 이 옵션은 기본적으로 비활성화 되어있으며 적은 비율의 샘플에서 비정상적으로 높은 수준으로 발현되는 유전자에 대해 결과가 풍부해 보이는 경우에만 권장됩니다.
- Statistical Comparison Tracks and Tables 의 Max Group Means 열에 이제 RPKM 대신 TPM이 표시됩니다. 이 열은 Create Heat Map for RNA-Seq 및 Ingenuity Pathway Analysis 플러그인 의 경로 분석 도구와 같은 도구에서 데이터를 필터링하는 데 사용됩니다.
Detect and Refine Fusion Genes
- 이전 Biomedical Genomics Analysis 플러그인에 배포되었던 Detect and Refine Fusion Genes의 업데이트된 버전입니다 .
- Fusion은 overlapping 유전자에 대해 calling 되지 않습니다.
- Novel exon boundary improvements:
- 옵션이 확장되어 Single fusion partner (“Detect with novel exon boundaries”)와 2 fusion partners (“Allow fusions with novel exon boundaries in both genes”)가 있는 융합을 탐지할 수 있습니다.
- “Detect exon skippings” 옵션은 새로운 Exon boundaries와의 fusions 감지를 지원합니다.
- 보고서에서 non-significant breakpoints를 생략하는 옵션이 추가되었습니다.
- Fusion에 대한 증거를 평가할 때 사용할 최소 Z-score를 지정할 수 있습니다.
- "Allow fusions with novel exon boundaries in both genes" 옵션이 false positive Fusion의 수를 줄이기 위해 기본적으로 false으로 설정됩니다. true로 설정하면 새로운 융합을 검색하는 데 유용합니다.
- Changes to the maximum number of equivalent matches to the reference allowed for a single read to be retained:
- Reads를 fusion chromosome으로 재 Mapping할 때 최대 수는 이제 30입니다. (이전에는 10이었습니다)
- unaligned ends를 검색할 때 최대 수는 변경되지 않고 10으로 유지됩니다.
- “Maximum number of hits for a Read” 옵션이 제거되었습니다.
- “Only use fusion primer Reads” 옵션이 활성화된 경우 paired end Reads가 single end Reads 으로 처리되는 문제를 수정했습니다.
- Insertions 및 deletions를 포함하는 Read에 대해 unaligned ends가 너무 길거나 너무 짧을 수 있는 문제를 수정했습니다. 따라서 이전 버전과 비교하여 결과에 약간의 차이가 발생할 수 있으며, 이는 결과의 false positive 와 false negatives의 감소로 인한 것으로 예상됩니다.
Bisulfite mapping
- Map Reads to Bisulfite Reference 툴의 분석 속도가 향상 되었습니다. (데이터 특성에 따라 속도가 다를 수 있습니다)
- Call Methylation Level 툴의 분석 속도가 향상 되었습니다.
- Import of Read mappings from SAM/BAM는 이제 선택적 SAM tags XR의 methylation 정보를 Read conversion에 사용하고 XG를 reference conversion에 사용합니다. 인식되는 값은 “CT” 및 “GA”입니다. Bisulfite Mapping 데이터를(SAM/BAM ) 내보내고 다시 가져오는 경우 정보가 손실되지 않도록 합니다.
- SAM/BAM파일의 Export시 bisulfite conversion에 대한 세부 정보가 포함됩니다.
- Read conversion을 위한 SAM tags XR과 reference conversion을 위한 XG를 사용하여 지정됩니다. tags의 가능한 값은 “CT” 및 “GA”이며, 이는 다른 툴들과의 호환성 향상을 위해 제공됩니다.
Workflows
- Branch on Coverage - 입력된 Coverage 범위 값을 기반으로 Mapping 이후 분석을 수행합니다.
- Import with Metadata - Raw data들에 Metadata Table을 Overlapping하는 템플릿입니다.
- Demultiplex Reads 요소를 포함하거나 Split Sequence List 요소를 포함하는 Workflow를 Batch 모드에서 실행할 수 있습니다.
- Workflow의 Demultiplex Reads elements에서 바코드를 미리 구성할 수 있습니다.
- Workflow Export elements는 AWS S3의 위치로 내보내도록 미리 구성할 수 있습니다.
Search for Reads in SRA
- Technical Reads 와 biological Reads를 함께 다운로드 할 수 있습니다.
- 다운로드 할 Read 와 Read의 구조 및 방향을 구성할 수 있습니다.
- Accession query field에 여러 개의 Accessions을 입력하면 각각 별도로 검색 됩니다.
- Sequence목록의 최종적인 크기는 더 이상 제공되지 않지만 공간 요구 사항에 대한 추가 정보가 메뉴얼에 추가 되었습니다.
- Troubleshooting에 대한 정보가 매뉴얼에 추가 되었습니다.
Read mappings
- Apple Silicon processors의 Read mapping 속도가 향상되었습니다. (Map Reads to Reference , RNA-Seq Analysis , Map Reads to Contigs 및 Map Bisulfite Reads to Reference)
- Stand-alone Read mappings 및 Read mapping tracks에서 deletions이 coverage 그래프와 Reads에서 강조 됩니다.
- Stand alone Read mappings 의 경우 “Match coloring” Side Panel에서 compactness level이 “Packed”으로 설정된 경우 Read에 적용된 색상을 표시합니다.
Import and export
- VCF Import: Inversions (<INV>), Insertions (<INS>), Deletions (<DEL>) and Tandem duplications (<TANDEM:DUP>)의 Symbolic alleles를 지원합니다. Sequence 정보를 포함하지 않거나 100,000bp보다 긴 Symbolic alleles는 Variant tracks 대신 Annotation tracks로 가져옵니다.
- 동일한 vcf 레코드에 인코딩된 multiple loci가 있는 variants에 대한 처리가 개선되었습니다.
- VCF Export: Insertions (<INS>), Deletions (<DEL>), Tandem duplications (<TANDEM:DUP>)에 대한 Symbolic alleles를 표현합니다. deletions를 제외하고 Annotation tracks 의 Variants는 항상 Symbolic alleles로 내보내집니다. Annotation tracks의 Deletions 및 지정된 크기 이상의 Variants도 Symbolic alleles로 내보냅니다. (기본 크기는 1000 bp이며, 이는 InDels < 1000 bp가 Symbolic alleles로 표현되어야 한다는 QCI Interpret 요구 사항에 해당합니다)
- PacBio의 HiFi Reads를 CLC내로 가져오는 것을 지원합니다.
- fastq 파일로 내보낼 때 Reads 길이가 524,288bp에서 16,777,216bp로 증가했습니다.
- SAM/BAM Mapping Files importer:
- 속도가 개선되었습니다.
- References 의 Circular flag가 나타납니다.
- “GFF3 Export” 에서 헤더의 대소문자를 유지합니다.
- Standard Import의 사용 기록 정보가 자세히 나타납니다. (예: “CSV Table importer”, “Fasta importer” 등)
- Standard Import 는 AWS S3 위치의 파일을 가져오는 데 사용할 수 있습니다.
- 이미지를 Bitmap 기반 형식으로 내보낼 때 지원되는 최대 픽셀 수를 초과하지 않도록 화면 해상도 및 고해상도 옵션이 제한됩니다.
Sequence Lists
- Checkboxes를 활성화 하여 Sequence Lists의 보기 내에서 Sequence를 선택할 수 있습니다. list는 표시 여부에 따라 정렬할 수 있으며 표시된 Sequence를 삭제할 수 있습니다.
- In the Annotation Table view, the following changes have been made to the right-click menu: 선택한 Annotation의 Sequence를 삭제할 수 있습니다.
- 선택한 Annotation이 있는 Sequence의 이름을 클립보드에 복사할 수 있습니다.
- GFF로 내보내는 옵션이 GFF3 형식으로 내보내집니다.
- Table view 에서 선택한 Sequence를 삭제할 수 있고 선택한 Sequence의 이름을 복사할 수 있습니다.
CLC Metadata Tables
- Batch mode에서 분석을 시작하거나 Iterate element로 워크플로를 시작할 때 연결된 데이터가 있는 CLC Metadata Tables을 input으로 직접 사용할 수 있습니다. Workflow를 시작할 때 Batch 단위의 기반이 되는 열을 지정할 수 있습니다.
- 다른 CLC Metadata Table 또는 Excel, CSV 또는 TSV 파일의 콘텐츠 추가를 수행하는 새로운 옵션이 포함됩니다. CLC Metadata Table의 행을 선택하여 새로운 CLC Metadata Table을 만드는 데 사용할 수도 있습니다.
- 데이터를 CLC Metadata Table에 연결하면 미리보기가 표시됩니다.
Other improvements
- UniProt에서 Sequences 검색이 업데이트되고 개선되었습니다. PubMed 항목에 대한 링크 및 더 많은 정보가 포함됩니다.
- Quick Search 및 Local Search 기능이 개선되었습니다.
- Batch mode에서 launching workflows with control flow elements (Iterate, Collect and Distribute)를 사용하여 Workflow를 시작할 때 Batch 단위의 개요가 조정되었습니다. 이름의 일부를 기반으로 요소를 포함하거나 제외하여 배치 단위의 내용을 조정할 수 있습니다.
- Low Frequency Variant Detection, Fixed Ploidy Variant Detection 또는 Basic Variant Detection 은 Local Realignment를 사용하여 realign된 mapping과 함께 guidance variant track과 함께 사용된 경우 partial insertions 부분이 감지 될 수 있었습니다. 이제 full insertion이 보고되려면 하나 이상의 개별적 Read 내에 있어야 합니다.
- QC for Targeted Sequencing
- 유전자당 coverage 통계를 확인할 수 있습니다.
- RNA-Seq 분석에서 생성된 Read Mapping 데이터 분석을 지원합니다.
- “Genome Reference Consortium_masking_hg38_no_alt_analysis_set”은 Reference Data Manager를 통해 reference element로 제공되며 “hg38_no_alt_analysis_set” genome sequence를 사용하는 reference sets의 일부입니다. Genome Reference Consortium 에서 정의한 영역이 포함되어 있어, 주로 U2AF1 유전자에 영향을 미치는 영역을 포함하여 잘못된 duplications를 제거하는 역할을 합니다. Map Reads to Reference Tool 과 함께 유용하게 사용할 수 있습니다.
- Annotate with Exon Numbers
- Annotation, Expression 및 Statistical Comparison Track에 Exon 번호를 추가할 수 있습니다.
- 하나의 transcript 또는 CDS 의 Exon으로 Annotation을 수행합니다.
- Custom Criteria 필터는 Statistical Comparison Track, Statistical Comparison Table, IsomiR Table 및 miRNA Seed Table을 필터링하는 데 사용할 수 있습니다.
- Demultiplex Reads has been updated to:
- 일치하는 Reads가 없는 barcodes를 보고 합니다.
- 기록에 barcodes 이름을 표시합니다.
- RNA-Seq 분석을 통해 생성된 Create Sample Reports 및 Combined Report 의 보고서(“Fragment counting statistics Table”)에 Mapping된 Read 비율이 포함됩니다.
- Create Sample Report 에서 설정된 threshold를 초과하는 Coverage 범위를 가진 대상 지역 위치의 백분율을 QC metric으로 사용할 수 있습니다.
- QC for Sequencing Reads 에서 long Read는 100,000bp까지만 처리합니다.
- Local Realignment는 더 이상 RNA-Seq Read mapping의 intron과 같은 coverage가 없는 영역으로 Read를 realigns 하지 않습니다.
- Remove Duplicate Mapped Reads 는 개선된 방법이 적용되어 paired end Read를 처리할 때 duplicate Read를 식별합니다. 따라서 약간 더 많은 Read가 중복으로 간주될 수 있습니다.
- Assemble Sequences to Reference은 circular reference의 원점에 걸쳐 있는 Read의 alignment를 지원합니다.
- Secondary Peak Calling 에는 Peak detection stringency 라는 새로운 옵션이 생겼습니다.
- The report from Copy Number Variant Detection (CNVs)
- CNV calling에 의해 영향을 받는 gene의 수를 보여주는 Table을 포함합니다.
- genome 및 chromosome 수준에서 새로운 coverage plot을 포함합니다.
- Trim Reads 보고서에는 이제 intact pairs 와 broken pair Read 수에 대한 통계가 포함됩니다.
- Restriction site database를 REBASE 2022-06-30으로 업데이트 했습니다.
- CLC Genomics Workbench용 Plug in은 Workbench가 Viewing Mode에서 실행 중일 때도 설치할 수 있습니다.
- 이외에 다양한 사소한 부분이 개선되었습니다.
Changes
- Toolbox의 RNA-Seq 및 Small RNA Analysis 폴더에 있는 Tool는 자연스러운 분석 흐름에 맞추어 하위 폴더로 정렬되었습니다.
- “Cloning tool” 은 “Restriction Based Cloning” 으로 이름이 변경 되었습니다.
- read mapping tracks 및 stand-alone read mappings 에 대한 “Disconnect paired reads” 옵션이 “Show strands of paired reads” 옵션으로 대체되었습니다.
- AWS Connections
- AWS connection 설정에서 AWS region을 지정할 수 있습니다. AWS connection이 이미 정의된 이전 버전에서 업그레이드하는 경우 region은 기본적으로 us-east-1로 설정되며 변경이 가능합니다. CLC Genomics Cloud 설정에서 실행되도록 CLC Cloud Module이 설치된 CLC Workbench에서 분석을 제출하려는 경우 필요합니다.