
ISMB / ECCB 2015 학회 참석기
- Posted at 2015/08/31 11:10
- Filed under 생물정보

ISCB(International Society for Computational Biology)에서 주최하는 ISMB(The 23rd Annual International Conference on Intelligent Systems for Molecular Biology)가 2015년 7월 11일부터 14일까지 4박 5일 동안 아일랜드 더블린의 Dublin Convention Centre에서 개최됐다. 이번 ISMB는 ECCB(The 14th Annual European Conference on Computational Biology)와 함께 열려 더욱 풍성한 내용을 담은 교류의 장이었다. ISMB와 ECCB는 bioinformatics와 computational biology, genomics, computational structural biology는 물론 system biology를 포함한 공통의 관심사를 갖는 국제학회이기 때문에 2004년부터 매 2년 마다 학회를 함께하고 있다. 2017년에 열릴 ISMB/ECCB는 체코의 수도인 프라하에서 열린다고 하니 국내외 연구자들과 세계적으로 아름다운 도시에서 지식을 나눈다는 것이 기대된다. 이번 ISMB/ECCB2015가 열린 아일랜드의 더블린 또한 역사와 전통이 깊은 도시로 많은 기대를 품고 참석했다.
-
ISMB
-
ISMB convenes an interdisciplinary group of scientists dedicated to the advancement of biological discovery through computation
-
ISMB educates scholars at all stages of their career
-
ISMB showcases state-of-the-art advances in the dynamic fields of computational biology and bioinformatics
-
ISMB is the forum for introducing new directions and for announcing technological breakthroughs
-
-
ISCB
- Leading professional society for computational biology and bioinformatics
- Connecting, Training, Empowering, Worldwide
Motivation
세기의 과학자 진 마이너의 예언 적중

작년(2014년) 미국 보스턴에서 열린 ISMB에서 진 마이너(Eugene Myers, Director and Tschira Chair of Systems biology, Max Planck Institute of Molecular Cell Biology and Genetics)는 "앞으로는 너희가(Bioinformatican) 직접 de novo assembly 할 필요 없는 시대가 올 것이다. 곧 시퀀싱 머신이 이 부분을 수행 할 것" 이라고 선언한 바 있다. 그런데 이와 맞물려 올해(2015년) 초 PacBio의 P6-C4 Chemical 기술발전과 더불어 전 세계적인 인기 상승이 시작됐다. 진 마이너가 PacBio의 개발상황을 알고 있었는지 모르겠지만, 이 선언은 실제로 현재 체감되고 있고 이 부분에 대한 자신의 견해를 Key note에서 공유한 것이다. (물론, de novo assembly 기술이 필요 없다는 것도 아니고 bioinformatican으로써 공부할 필요가 없다는 것도 아니다. 다만, 시퀀싱머신에서 이를 수행할 것이라는 예견일 뿐이다.) 진 마이너의 정확한 안목을 확인하며 개인적으로 엄청난 도전의식을 받았고 국제학회의 중요성을 인식했다. 따라서 이번 ISMB2015에서도 이러한 동향을 확인하는 것에 초점을 맞추고, 최신 분석 pipelines과 tools을 공부해 직접 적용이 가능한 최신의 그 무언가를 얻고자 하는 마음가짐으로 이번 학회에 참석했다.
Attendance
ISMB를 즐기는 방법
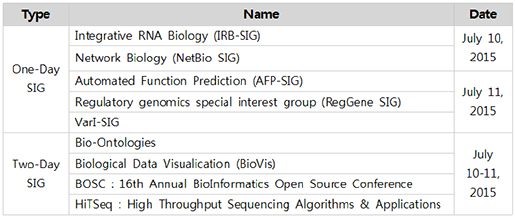
5일 동안 진행되는 이번 ISMB/ECCB2015의 일정은 크게 pre-conference(July 10-11, 2015)와 main-conference(July 12-14, 2015) 두 가지로 나뉜다. Pre-conference 기간 동안에는 SIGs(Special Interest Groups) 미팅이 진행되며 이 미팅은 총 9개의 세부 모임으로 구성된다(Table 1). 각각의 모임은 등록기간 내에 신청하고 일정량의 금액을 미리 지불해야한다. 이번 학회의 SIGs 모임은 BioVis와 HiTSeq을 신청해 참가했는데 BioVis에서는 주로 pathway, GO, comparative genomics에 대한 visualization을 다뤘고, HiTSeq 에서는 NGS를 통한 다양한 응용연구분야에 대한 톡이 주를 이뤘다.
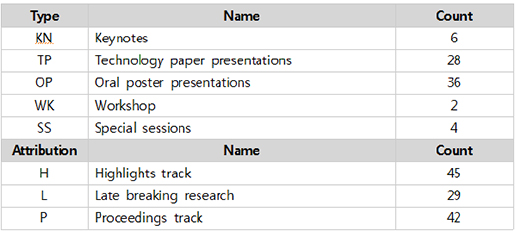

또한, 각 세션마다 테마를 설정해 학회 참석자들에게 선택의 편의성을 제공한 점은 작년(2014년)과 다른점이다. 테마는 GENES, DISEASE, PROTEIN, SYSTEMS, DATA, OTHERS로 구성되어 있다.
- DATA : Includes data and text-mining, ontologies, databases and machine learning approaches that do not fit in other categories.
- DISEASE : Includes analysis of mutations, phenotypes, drugs, epidemiology and other clinically relevant areas.
- GENES : Includes work in genes (including non-coding RNA), transcriptomes, genomes and variation.
- PROTEINS : Includes analysis of proteins and their structures and proteomics.
- SYSTEMS : This theme includes higher level systems such as cells, tissues, whole organisms and ecosystems. Includes systems biology, molecular interactions and genetic regulation.
- OTHERS : Research areas that do not fall within the five (5) main thematic areas. The organizers may, at their discretion, move submissions to other thematic areas.
July 12-13 저녁시간에는 저자와 학회 참석자가 함께 자유로운 토론을 할 수 있는 포스터리셉션이 진행됐다. (주)인실리코젠도 non-model species analysis를 주제로 두 편의 포스터와 함께 참가했다. ISMB/ECCB 학회의 특성상 system biology 분야가 주를 이루기 때문에 non-model species analysis에 대한 큰 관심을 기대하지 못했다. 하지만 많은 국외연구자들이 non-model species에 대한 관심을 갖고 있었고 non-well model species의 re-annotation에도 큰 관심을 갖고 있었다. 질의 중 새로운 관점을 느꼈는데 transcriptome analysis에서 de novo assembly와 expression abundance를 계산할 때 샘플링 단계에서 total mRNA를 취할 것이냐, single cell을 취할 것이냐에 대한 것이었다. 최근 분석 트렌드는 single cell에서의 development 등을 확인하는 것인데 그것을 염두한 의견같다. Single cell이 pooling cell을 커버할 수 있을 것인가와 pooling cell에서 missing point가 생기지 않을까에 대해 나눴고, 보고자 하는 연구 목적에 따라 다르다는 결론을 내렸다.
참가 포스터 1-
제목 : An integrated pipeline and monitoring system for de novo genome analysis [F09]
-
저자 : Junhyung Park, SeungJae Noh, Kyuyeol Lee, Yeonkyung Kang, Myunghee Jung

-
제목 : De novo transcriptome assembly and in silico expression PROFILES of Sebastes schlegeli [E41]
- 저자 : Seung Jae Noh, Sathiyamoorthy Subramaniyam, Seungil Yoo, Jehee Lee, Jae-Koo Noh, Bohye Nam
Main-conference 기간 중에는 booth exhibitors를 통해 정보를 얻을 수 있었다. (주)인실리코젠의 국내외 협력업체 중 하나인 QIAGEN Bioinformatics도 이번 ISMB/ECCB2015에 참석해 자리를 빛냈다. QIAGEN은 미국 메사추세스주 비벌리에 위치해 있으며 NGS를 이용한 bioinformatics software tools을 서비스하고 있다. 최근 CLC bio, Ingenuity, BIOBASE 사를 합병해 더욱 다양한 분야의 분석과 우수한 DB를 바탕으로 통합분석의 발판을 마련하고 있는 중이다.
 Figure 2. Exhibitors of ISMB/ECCB2015
Figure 2. Exhibitors of ISMB/ECCB2015
Figure 3. With Qiagen
ISMB에서는 현재...
최신분석기법 및 도구
- LINKS
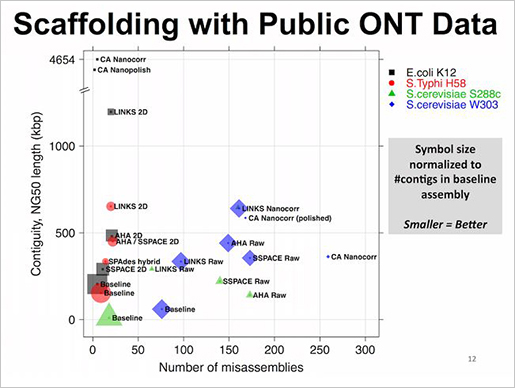
현재까지 공개된 scaffolding 도구들과 비교를 했을 때도 mis-assemblies가 적고 contiguity나 NG50길이가 긴 것을 확인 할 수 있다.
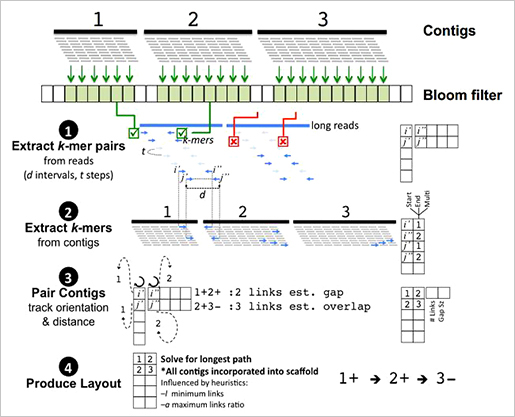
LINKS는 메모리 효율성이 매우 뛰어나다. 그 이유는 scaffolding algorithm에 있다. long reads를 짧은 k-mer pairs로 추출하고 scaffolding의 대상이 되는 contigs도 k-mer pairs를 추출한다. 각각에서 추출된 k-mer pairs의 서열상동성이 같은 위치정보와 paired-end information을 통해 scaffolding을 수행한다. 또한 iteration 수를 높게 조절함으로써 정확도를 향상할 수 있다는 장점도 존재한다.
- BactoGeNIE

위 사진은 E.coil의 700 strains에 대해서 neighborhood around a hypothetical protein을 확인하는 장면이다. Display는 at 21.9 by 6.6 feet and 6144 by 2304 pixcels이다.
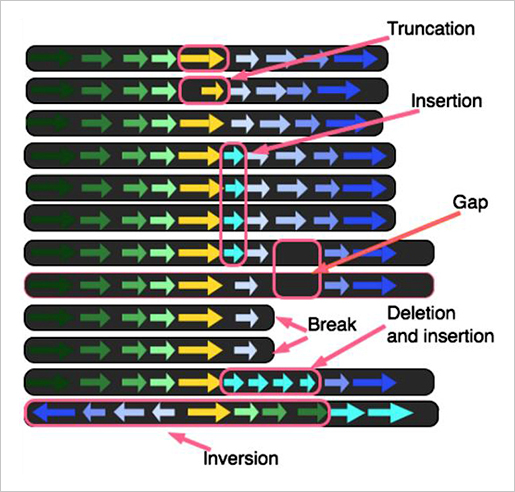
각각의 genome은 한 행에 하나씩 배열되며 배열된 모든 genome의 alignment를 통해 comparative genome analysis를 수행한다. 각각 유전체의 특징적 단위가 화살표로 표시되며 breaks, deletions, insertions, gaps 등을 확인 할 수 있다.
- Clustal Omega

Clustal Omega는 최초 guide-trees를 생성하기 위해 mBed (Blackshields, 2010) calculates distance matrix를 채택했다. 이로써 기존에 large (N > 10,000) alignments distance matrix가 갖는 bottleneck을 해결했다. Fabian Sievers(University College Dublin)의 말에 따르면, Clustal 시리즈의 고질적 문제였던 ‘any size'의 alignment가 가능해졌고, 퍼포먼스 또한 크게 좋아졌다.
데이터베이스
-
UniProt

데이터의 내용을 보호하면서 사이즈만을 어떻게 줄일 것이냐? 라는 질문에 가장 중요한 단계는 sequence comparison이라 답했다. CD-Hit-2D를 사용해 두 세트의 서열을 비교했으며 90%이상의 sequence identity threshold와 90%이상의 proteome similarity threshold로 서열상 redundancy를 제거했다.
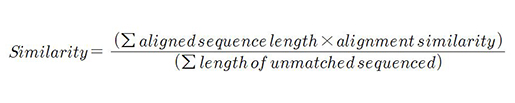
매우 많은 entry가 줄어서 데이터베이스의 크기 자체는 줄었지만 훨씬 specific하고 meaningful한 데이터베이스가 탄생했다. 줄어든 데이터베이스는 ordering을 통해 여러 개의 component로 merge되어있다. 현재 UniProt site에 released database는 proteome redundancy removal 버전이다.
- EVA

주요 관심사 및 최신동향
- Single cell RNAseq studies
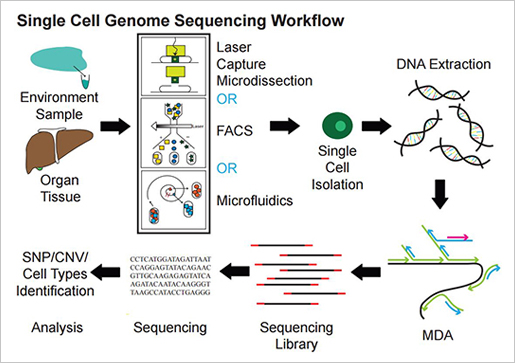
국내 연구동향 및 시퀀싱 회사의 single cell analysis는 아직 걸음마 단계에 불과하다. Single cell에서 DNA와 RNA를 분리해 시퀀싱하고 그 안에서 일어나는 생물학적 이벤트는 epigenomics 연구는 물론, cell cycle에 따른 cell-to-cell correlations, T cell 연구 등 다양한 분석에 적용이 가능하다.
- GBS
Genotyping By Sequencing (GBS)는 차세대 시퀀싱 기술을 바탕으로 새롭게 개발, 발전하고 있는 NGS 분석법 중에 하나이다. 유전체 전체를 시퀀싱하는 WGS에 비해 저렴한 비용으로 빠르고 쉽게 genome-wide 분석을 가능하게 한 테크닉이다. 제한효소를 처리하여 유전체 서열에서 그 제한효소에 의해 잘리는 영역 주변의 서열만을 시퀀싱하게 된다. 제한효소를 처리한다는 점에서는 RAD-seq과 근본적으로 원리가 같지만 효소절단 후 사이즈 선별을 하지 않는다는 점에서 시퀀싱 라이브러리 제작이 보다 간단한 편이며 GBS 시퀀싱 데이터가 RAD-seq에 비해 low coverage로 얻어진다.
GBS는 아래와 같은 applications이 있다.
- Marker discovery
- Phylogeny/Kinship
- Linkage mapping of QTL in a biparental cross
- Fine mapping QTL (Quantitative trait loci)
- Genomic selection
- GWAS (Genome wide association study)
- NAM-GWAS (Nested association mapping GWAS)
- Improving reference genome assembly

Impression
이젠 국내에서도 bioinformatics의 인식이 달라져야 한다.
작년 ISMB2014에서는 de novo assembly 나 expression analysis, GO, KEGG, COG, pathway 연구와 같은 전반적이고 일반적인 연구가 상당수를 차지했다. 하지만 올해의 ISMB학회는 ECCB와 함께했기 때문인지 그런 비율이 많이 줄었고 좀 더 세분되고 집중적인 연구가 주를 이뤘다. 특히 암과 같은 특정 질병의 원인 판별과 치료법 개발에 초점이 맞춰져 있었다. 세션들의 주제가 GENES, PROTEINS, SYSTEMS, DATA 이외에 DESEASE도 있다는 것은 이를 반증하고 있으며, DESEASE 세션의 개수도 상당하고 그 연구수준도 점진적으로 발전되고 있어 보인다. 특히 연구방법들이 한가지 방향으로 모이지 않고 다양한 시도를 통한 기초연구에 힘쓰는 것에 놀랐다. 이는 한국의 연구풍토와 유럽, 미국의 연구풍토가 많이 다르기 때문으로 생각한다. 언제쯤인지 퇴근길에 읽은 어느 연구자님의 문구에 따르면, 정확한 메커니즘의 이해보다는 응용성을 강조하는 우리나라의 스타일과는 다르게 유럽이나 미국은 정확한 원리를 이해하고 그것을 수학적인 도구를 통해 분석해야 하며 실험의 결과들이 높은 재현성을 나타냄과 더불어 각각의 결과들이 강한 유기성을 가질 때 비로소 올바른 결과로써 인정한다고 한다. "블루오션은 찾아내는 것이 아닌 만드는 것이다.", "소비자는 자신이 무엇을 원하는지 모른다."는 명언들이 말하는 창조적 사고의 연구풍토가 선진국이 될 한국에도 자리 잡을 때가 아닌가 생각한다.
NGS의 도입과 함께 전체를 아우르는 분석이 가능해졌고 새로운 그 무언가를 찾을 기회가 많아졌다. 작년까지는 이런 NGS의 장점과 특성을 살린 연구추세였다면 점차 NGS의 응용범위가 좁은 범위까지 확대되어가는 중이다. Iontorront에 이어 nanopore와 같은 소형 시퀀싱머신의 개발과 보급이 일반화되기 시작했다. 앞으로는 국내의 실험실에도 많은 변화가 있을 것이며, 그 중심은 또 한 번 NGS가 될 것이다. 이젠 국내에서도 bioinformatics의 인식이 달라져야 한다.
유승일 컨설턴트
- Introduction
- Clustal omega
- Uniprot
- EVA
- GBS
- Single cell analysis
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/188
Trackback URL : 이 글에는 트랙백을 보낼 수 없습니다

