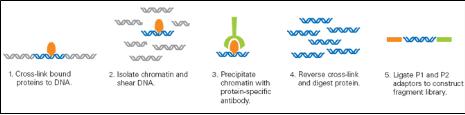
BKL PROTEOME
- Posted at 2010/05/04 16:34
- Filed under 제품소식
진핵생물의 세포내 조절 메카니즘은 전사 수준의 유전자 발현 조절과 이후 생성된 단백질 간의 조절 메카니즘으로 구분지어 볼 수 있다. 이들은 세포 밖 외부 신호로부터 target 유전자까지의 신호전달을 유기적으로 전달하며 다양한 루트를 통해 전달한다. 따라서 하나의 단백질과 유전자가 한 가지 기능만을 수행하기 보다는 다양한 단백질과 유전자들과의 상호 협력적인 관계를 통해 전체적인 세포내 항상성을 유지하게 된다.
Biobase는 이러한 총체적인 세포내 조절 메카니즘 분석을 위해 BKL TRANSFAC을 통해 전사수준의 세포내 조절 메카니즘 분석을 위한 resource 데이터를 제공하고, BKL PROTEOME을 통해 이후 단백질 수준의 조절 메카니즘 분석을 위한 데이터베이스를 서비스하고 있다.
2010년 현재 PROTEOME은 6개의 category로 구분된 데이터베이스로 운영되고 있다. Disease-biomarker associations 관심 있는 유전자 혹은 질병에 관련된 pathway, regulation networks, drug interaction 정보를 제공하며 단백질과 질병간의 조절관계를 모 식도를 통해 이해하기 쉽도록 다양한 정보를 제공하고 있다.
Drug-protein interactions 특정 약물에 의해 영향을 받는 대사회로 및 네트워크 정보를 제공하며, 이는 drug 개발을 위한 결정에 보다 직관적인 정보를 제공 한다.
Signaling, metabolic pathway, and expression regulation data 모식화 된 pathway 및 regulation networks 정보를 통해 세포내 조절 메카니즘을 총체적으로 이해 할 수 있도록 하였다.
Plant Science public data와 전문가의 manual curation 데이터의 조합을 통한 식물 유전체 내의 pathway 정보와 resource data를 제공한다.
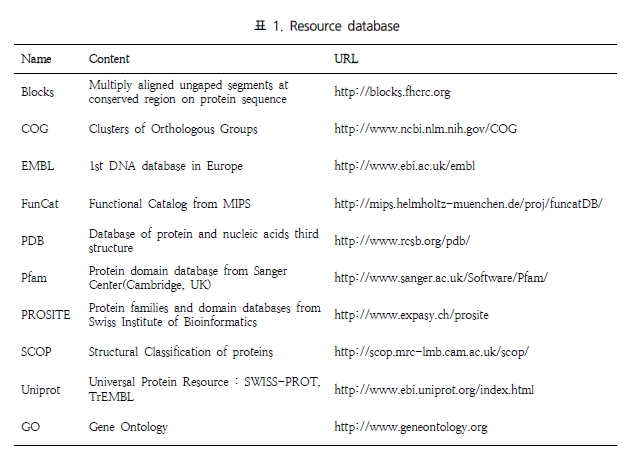
Quick search BKL PROTEOM은 Gene/protein, disease, pathway, drug 그리고 keyword category를 통해 검색 할 수 있다. 원하는 유전자가 포함된 disease 및 pathway정보를 문헌을 통한 전문가의 curation으로 세포내 기능을 검색할 수 있다.
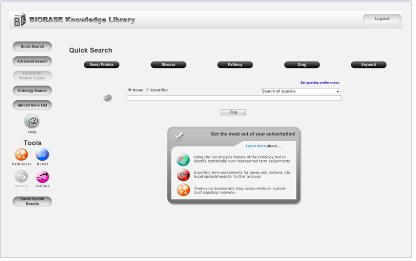 Figure 1. BKL PROTEOM quick search. 유전자, 질병, pathway, drug, keyword를 통해 원하는 정보를 손쉽게 검색할 수 있다. 또한 organism을 제한하여 많은 데이터들 속에서 원하는 정보만을 한 번에 검색 할 수 있다.
Figure 1. BKL PROTEOM quick search. 유전자, 질병, pathway, drug, keyword를 통해 원하는 정보를 손쉽게 검색할 수 있다. 또한 organism을 제한하여 많은 데이터들 속에서 원하는 정보만을 한 번에 검색 할 수 있다.
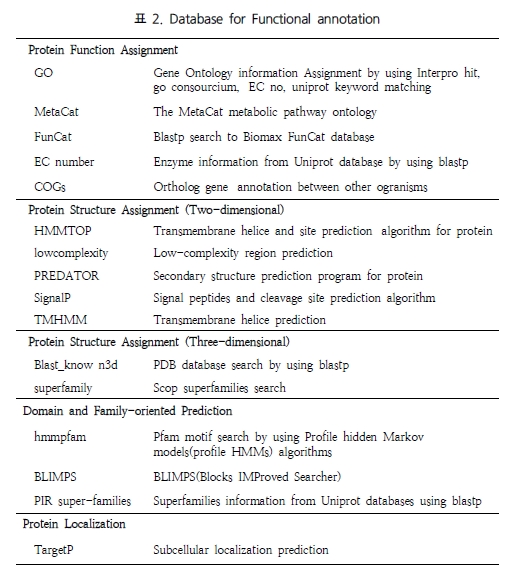
STAT3 단백질을 검색한 결과 기본적인 단백질의 대표 기능과 함께 다양한 데이터베이스에서 활용되고 있는 STAT3의 synonyms 정보를 서비스 한다. 또한 좀 더 세분화된 카테고리로 구분된 단백질의 정보를 서비스하는데, biomarker
associations, drug interaction, gene ontology, mutant phenotype, pathway, transcriptional regulation, protein feature, annotation에 관련된 세포내 총체적인 기능을 이해 할 수 있도록 서비스 하고 있다.
Biomarker association disease와 관련된 biomarker로 활용되고 있는 단백질의 정보를 서비스한다. 이러한 정보는 질병의 진단을 위해 혹은 질병 징후에 대한 연구를 위해 활용되고 있다(Figure 2).
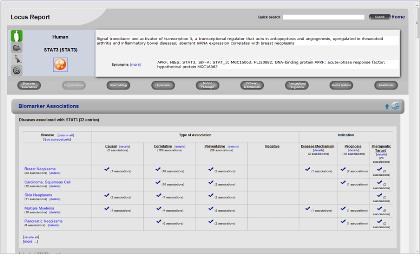 Figure 2. Biomarker association. 질병과 관련한 단백질의 표지인자로 활용되는 정보를 서비스한다. 각 질병과 관련된 단백질의 상세 관계 정보는 질병을 클릭하여 자세히 확인 할 수 있다.
Figure 2. Biomarker association. 질병과 관련한 단백질의 표지인자로 활용되는 정보를 서비스한다. 각 질병과 관련된 단백질의 상세 관계 정보는 질병을 클릭하여 자세히 확인 할 수 있다.
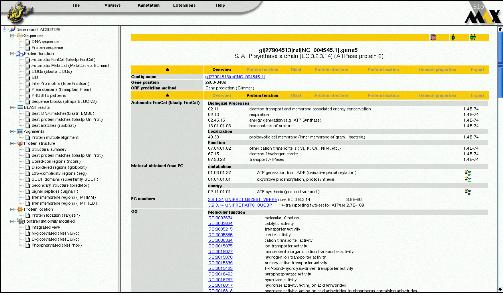
Pathway interaction 단백질과 관련한 pathway 및 interaction 정보를 서비스 한다(Figure 3). Multi-function하는 단백질의 특성상 다양한 pathway와 interaction정보를 검색 할 수있으며 이들의 pathway는 모식도를 통해 graphical하게 확인 할 수 있다. Pathway상의 upstream, downstream에 존재하는 단백질과 관계정보를 총체적으로 살펴 볼 수 있으며 이들 정보는 모두 text 형태로도 변환이 되어 서비스된다.
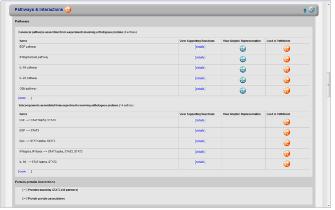 Figure 3. Pathway & Interaction. 관심 있는 단백질이 포함된 pathway와 interaction정보를 모식도를 통해 서비스하고 있다.
Figure 3. Pathway & Interaction. 관심 있는 단백질이 포함된 pathway와 interaction정보를 모식도를 통해 서비스하고 있다.
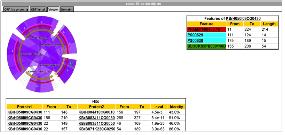
Pathway 모식도는 figure 4에서 보여지는 것과 같이 대표 단백질로 간편화 하여 전
체적인 세포내 기능을 이해 할 수 있는 것(figure 4. A)과 관련 단백질의 모든 관계를 표시한 PathFinder(figure 4. B)로 구분 지어 있다. PathFinder는 많은 단백질의 관계 중에 보고자하는 특정 질병 및 drug 그리고 유전자 관련 pathway만을 지정하여 tag를 이용하여 표시함으로써 이해를 돕고 있다.
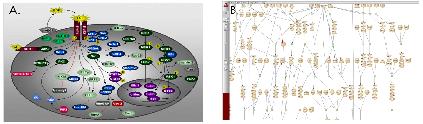 Figure 4. Pathway. Graphical viewer를 통한 pathway의 주요 단백질 만들 대상으로 전체적인 정보를 보여주는 것(A)과, PathFinder(B)를 통한 모든 관련 단백질의 관계를 포함한 질병 및 drug 정보를 자세히 살펴 볼 수 있다. PathFinder에서는 zoom-in/out을 통해 단백질간의 관계를 자세히 살펴 볼 수 있으며, 원하는 단백질, 질병, drug정보를 기준으로 직접적으로 영향을 주는 pathway에 하이라이트를 통해 보다 직관적으로 이해할 수 있도록 하였다.
Figure 4. Pathway. Graphical viewer를 통한 pathway의 주요 단백질 만들 대상으로 전체적인 정보를 보여주는 것(A)과, PathFinder(B)를 통한 모든 관련 단백질의 관계를 포함한 질병 및 drug 정보를 자세히 살펴 볼 수 있다. PathFinder에서는 zoom-in/out을 통해 단백질간의 관계를 자세히 살펴 볼 수 있으며, 원하는 단백질, 질병, drug정보를 기준으로 직접적으로 영향을 주는 pathway에 하이라이트를 통해 보다 직관적으로 이해할 수 있도록 하였다.
Regulation 특정 단백질이 조절하는 다른 단백질 정보를 서비스하는 것으로 up-regulation, down-regulation 그리고 non-effect로 구분되어 있다(figure 5). Pathway상에서 찾아 볼 수 있는 정보를 보다 유연한 형태로 서비스함으로써 사용자 편의를 고려한 서비스라 하겠다.
 Figure 5. Regulation. 단백질들 간의 조절 관계를 up-/down-regulation을 통해 정리하였다.
Figure 5. Regulation. 단백질들 간의 조절 관계를 up-/down-regulation을 통해 정리하였다.
Annotation 단백질의 pathway 정보뿐만 아니라 expression정보, GO 정보, modification 정보, localization 정보를 비롯한 단백질의 모든 기능을 서비스한다. 이들 정보는 모두 전문가의 curation을 통해 정리된 것으로 참고가 된 문헌 정보는 모두 링크를 통해 서비스 되고있다(figure 6).
 Figure 6. Annotation. 단백질의 다양한 function 정보를 서비스한다. Pathway를 비롯한 expression, domain, GO, mechanism, feature정보를 문헌정보와 함께 서비스하고 있다.
Figure 6. Annotation. 단백질의 다양한 function 정보를 서비스한다. Pathway를 비롯한 expression, domain, GO, mechanism, feature정보를 문헌정보와 함께 서비스하고 있다.
2010년 4월 업데이트를 통해 새롭게 서비스 되는 BKL PROTEOM은 이전 버전과 비교하여 사용자 편의를 고려한 서비스가 매우 강화 되었다. Export 기능을 통해 원하는 정보를 모두 다운로드 받을 수 있으며, 많은 정보 가운데 원하는 정보만을 선택적으로 살펴 볼 수 있도록 카테고리화 한 점도 이에 해당한다. 그러나 무엇보다 Biobase의 최대 장점은 문헌정보를 바탕으로 한 전문가의 curation으로 데이터의 신뢰성을 높였다는 것으로 BKL PROTEOM 또한 신뢰성 높은 데이터베이스를 제공하고 있다.
Biobase는 이러한 총체적인 세포내 조절 메카니즘 분석을 위해 BKL TRANSFAC을 통해 전사수준의 세포내 조절 메카니즘 분석을 위한 resource 데이터를 제공하고, BKL PROTEOME을 통해 이후 단백질 수준의 조절 메카니즘 분석을 위한 데이터베이스를 서비스하고 있다.
2010년 현재 PROTEOME은 6개의 category로 구분된 데이터베이스로 운영되고 있다. Disease-biomarker associations 관심 있는 유전자 혹은 질병에 관련된 pathway, regulation networks, drug interaction 정보를 제공하며 단백질과 질병간의 조절관계를 모 식도를 통해 이해하기 쉽도록 다양한 정보를 제공하고 있다.
- 자연계에서 일어나는 현상에 대한 인과 관계 및 예방을 위한 정보
- mRNA의 과잉 발현, DNA mutation, altered protein의 activity와 관련된 질병정보
- 해당 약물의 질병 메카니즘에 끼치게 될 영향 및 target 유전자에 가해질 잠재적인 예후 정보
- 새롭게 찾아낸 단백질의 다양한 pathway 정보 및 관계 정보
Drug-protein interactions 특정 약물에 의해 영향을 받는 대사회로 및 네트워크 정보를 제공하며, 이는 drug 개발을 위한 결정에 보다 직관적인 정보를 제공 한다.
- Drugbank 로부터 7,000개의 drug-protein interaction 정보를 분석
- Yeast에서 확인된 1,200개의 chemical regulation 정보
- Human, yeast 그리고 worm에서 annotation된 12,000개의 drug interaction의 자세한 정보.
Signaling, metabolic pathway, and expression regulation data 모식화 된 pathway 및 regulation networks 정보를 통해 세포내 조절 메카니즘을 총체적으로 이해 할 수 있도록 하였다.
- 19,000 건의 signaling interactions
- Fungal 유전자의 2,700개의 regulator정보
- 5,100건의 pathway 정보
- S.cerevisiae, S.pombe - 질병, 노화, fungal pathogen, 바이오연료 그리고 그 외 기초 연구를 위한 모델 정보
- C.elegans - 질병, 노화, miRNA technology, nematode pathogen 그리고 그 외 기초 연구를 위한 모델 정보
- C. albicans and other Candida species
- Aspergillus species
- Blastomyces species
- Coccidioides immitis
- Cryptoccocus neoformans
- Histoplasma capsulatum
- Pneumocystis species
Plant Science public data와 전문가의 manual curation 데이터의 조합을 통한 식물 유전체 내의 pathway 정보와 resource data를 제공한다.
- Arabidopsis, soybean, maize, sorghum, and rice
- 다른 데이터베이스에서는 찾아 볼 수 없는 표현형과 발현치에 대한 정보
- Cell signaling and metabolic pathway data
- BAR을 통한 발현데이터 visualization
- Sequence 정보를 이용한 규명되지 않은 단백질의 GO, domain정보
PROTEOME Tutorial
Quick search BKL PROTEOM은 Gene/protein, disease, pathway, drug 그리고 keyword category를 통해 검색 할 수 있다. 원하는 유전자가 포함된 disease 및 pathway정보를 문헌을 통한 전문가의 curation으로 세포내 기능을 검색할 수 있다.
STAT3 단백질을 검색한 결과 기본적인 단백질의 대표 기능과 함께 다양한 데이터베이스에서 활용되고 있는 STAT3의 synonyms 정보를 서비스 한다. 또한 좀 더 세분화된 카테고리로 구분된 단백질의 정보를 서비스하는데, biomarker
associations, drug interaction, gene ontology, mutant phenotype, pathway, transcriptional regulation, protein feature, annotation에 관련된 세포내 총체적인 기능을 이해 할 수 있도록 서비스 하고 있다.
Biomarker association disease와 관련된 biomarker로 활용되고 있는 단백질의 정보를 서비스한다. 이러한 정보는 질병의 진단을 위해 혹은 질병 징후에 대한 연구를 위해 활용되고 있다(Figure 2).
Pathway interaction 단백질과 관련한 pathway 및 interaction 정보를 서비스 한다(Figure 3). Multi-function하는 단백질의 특성상 다양한 pathway와 interaction정보를 검색 할 수있으며 이들의 pathway는 모식도를 통해 graphical하게 확인 할 수 있다. Pathway상의 upstream, downstream에 존재하는 단백질과 관계정보를 총체적으로 살펴 볼 수 있으며 이들 정보는 모두 text 형태로도 변환이 되어 서비스된다.
Pathway 모식도는 figure 4에서 보여지는 것과 같이 대표 단백질로 간편화 하여 전
체적인 세포내 기능을 이해 할 수 있는 것(figure 4. A)과 관련 단백질의 모든 관계를 표시한 PathFinder(figure 4. B)로 구분 지어 있다. PathFinder는 많은 단백질의 관계 중에 보고자하는 특정 질병 및 drug 그리고 유전자 관련 pathway만을 지정하여 tag를 이용하여 표시함으로써 이해를 돕고 있다.
Regulation 특정 단백질이 조절하는 다른 단백질 정보를 서비스하는 것으로 up-regulation, down-regulation 그리고 non-effect로 구분되어 있다(figure 5). Pathway상에서 찾아 볼 수 있는 정보를 보다 유연한 형태로 서비스함으로써 사용자 편의를 고려한 서비스라 하겠다.
Annotation 단백질의 pathway 정보뿐만 아니라 expression정보, GO 정보, modification 정보, localization 정보를 비롯한 단백질의 모든 기능을 서비스한다. 이들 정보는 모두 전문가의 curation을 통해 정리된 것으로 참고가 된 문헌 정보는 모두 링크를 통해 서비스 되고있다(figure 6).
2010년 4월 업데이트를 통해 새롭게 서비스 되는 BKL PROTEOM은 이전 버전과 비교하여 사용자 편의를 고려한 서비스가 매우 강화 되었다. Export 기능을 통해 원하는 정보를 모두 다운로드 받을 수 있으며, 많은 정보 가운데 원하는 정보만을 선택적으로 살펴 볼 수 있도록 카테고리화 한 점도 이에 해당한다. 그러나 무엇보다 Biobase의 최대 장점은 문헌정보를 바탕으로 한 전문가의 curation으로 데이터의 신뢰성을 높였다는 것으로 BKL PROTEOM 또한 신뢰성 높은 데이터베이스를 제공하고 있다.
Posted by 人Co
- Tag
- annotation, BKL PROTEOME, BKL TRANSFAC, disease-biomarker, Drugbank, Human, insilicogen, mutation, PathFinder, pathogen, pathway, Signaling, STAT3, Yeast, 문헌정보, 인실리코젠
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/72