
R을 이용해 벤다이어그램(venn diagram) 그리기
- Posted at 2013/11/01 17:41
- Filed under 생물정보
<세상엔 예쁜 벤다이어그램이 참 많지요.. 저걸 일일이 언제 다 그리나... 하아...>
벤다이어그램을 그리는 방법은 어렵지 않습니다. 엑셀의 필터 기능을 이용해 공통집합들에 해당하는 유전자(원소) 수와 각 집합에 유니크한 유전자 수를 각각 알아낸 다음 파워포인트로 타원을 이리저리 교차 시켜 템플릿을 만들고, 겹치는 부분에 해당하는 숫자를 입력하기만 하면 됩니다. 하지만 언제나 그렇듯 우리의 데이터는 수만의 유전자와 최소 3개 이상의 샘플(혹은 그룹)이 있으며 여러가지 실험 조건에 따라 값이 변경된 다양한 버전들로 존재합니다. 게다가 그룹이 4개인 벤다이어그램은 부분집합 수만 16개인데, 그 이상의 그룹을 벤다이어그램으로 그리려면 그룹이 기하급수적으로 늘어나고, 이렇게 번거로운 작업을 매번 반복하기란 여간 귀찮은일이 아닙니다.
<그룹이 5개면 부분집합만 31개.... 엑셀로 하면 부분집합 필터링 하는데만 마우스 클릭만 몇 번을 해야 하나....>
그래서 데이터 테이블만 있으면 자동으로 공통집합의 원소수를 알아내 예쁜 벤다이어그램으로 그려주는 방법을 알아보고자 합니다.
여기서는 통계 계산과 그래픽에 많이 사용되는 R이라는 프로그래밍 언어를 사용해 보겠습니다. 이미 다양한 경로로 R에 대해 들어 보셨을 겁니다. 하지만 많은 연구자들이 R을 사용하는데 많은 장벽을 느끼고 좌절을 하곤 합니다. 저 역시 마찬가지였고요. 하지만 이런 기회를 통해 조금씩 R과 친해져 보는 것도 앞으로 연구를 계속 하는데 많은 도움이 될 것 같습니다. 사실 엑셀과 R이 하는 일은 크게 다르지 않습니다. 두려움과 귀찮음을 잠시 묻어두고 한걸음 나아가봅시다.R 설치하기
R은 R-Project(http://www.r-project.org/) 웹 페이지의 왼쪽 메뉴에서 'CRAN' =>
'http://cran.nexr.com/' 또는 'http://biostat.cau.ac.kr/CRAN/' (Korea의 미러사이트 중 한군데) =>'Download R for Windows' => 'base' => 'Download R 3.0.2 for Windows' 순서로 클릭을 하면 설치 프로그램을 다운로드 받을 수 있습니다. 다운로드가 완료되면 설치 프로그램을 실행하시고 아무 걱정 없이 '다음'을 쭉 클릭하시면 설치가 완료됩니다. 설치가 완료되면 '시작' => '모든 프로그램' => 'R' => 'R x64 3.0.2'을 마우스 우클릭을 하여 '관리자 권한으로 실행'을 클릭합니다. 32비트 컴퓨터를 사용하시면 'R i386 3.0.2'을 클릭합니다.
Data format, always data format
먼저 벤다이어그램을 그릴 데이터를 준비합니다. 모든 분석 코드들은 정해진 포맷에 맞는 데이터가 입력되어야만 에러 없이 결과를 출력합니다. 분석 코드의 첫부분은 입력 데이터의 어디가 이름(ID, valiable name)이고 어디가 값(value)인지, 어떻게 구분되어 있는지(tab-delimited, comma-separated)를 컴퓨터에게 인지시켜주는 내용들인데, 정해진 포맷의 데이터가 입력되지 않으면 엉뚱한 위치에서 값을 찾아 결국엔 에러 메세지를 출력합니다. 입력된 값에 이상이 있어 에러가 나기도 하지만 대부분의 에러는 입력데이터의 포맷이 사용한 코드에 맞지 않아 발생합니다. 이런 에러를 피하려면 두 가지 방법이 있습니다. 내 데이터 포맷을 분석 코드가 인식할 수 있도록 분석코드를 수정하는 겁니다. 하지만 이런 방법은 쉽지 않습니다. 두 번째 방법은 분석 코드가 인식하는 데이터 포맷에 맞게 내 데이터를 준비하는 겁니다. 준비 단계에서 약간 번거로울 수 있지만 가장 확실한 방법입니다.
먼저 실습해 볼 예제 데이터는 몇몇 근연종에서 ortholog gene을 분석한 데이터입니다.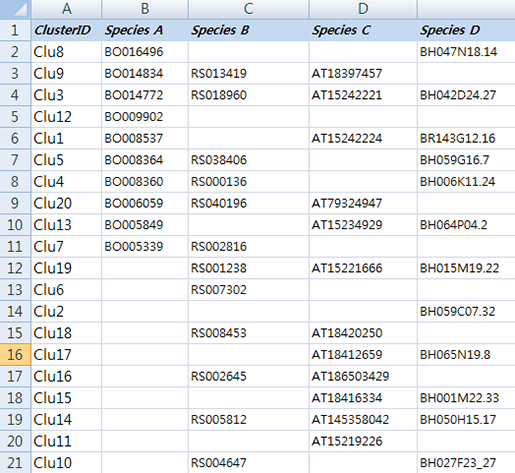
파일을 다운 받아 주세요.
 ortholog_example.txt
ortholog_example.txt이 데이터는 각 종의 유전자 서열을 서로서로 BLAST를 돌려 서로 일치하는 유전자들을 각 종의 gene ID로 표기한 테이블입니다. 예를 들어 Species A의 BO16496은 Species D의 BH047N18.
14와 ortholog 관계가 있다고 보여져 한
라인에 각각의 아이디를 표기 하고 Clu8이라는 ID를 부여했습니다. Clu3에 있는 유전자들은 모든 종에서 발견된
ortholog gene이네요. Clu6의 RS007302는 다른 종의 서열에서 일치하는 유전자가 없어 Species B에서의
specifie gene으로 보여집니다. (이 데이터는 예제 데이터로 ID와 ortholog 관계를 무작위로 작명하고 배열한 데이터입니다.)
패키지 불러오기
데이터가 준비 되었으면, 분석 코드가 사용할 패키지 코드를 설치합니다. 패키지는 어떤 분석에 주로 사용되는 분석 코드를 기능 별로 만들어 모아 놓은 것을 말합니다. 여기서는 limma라는 패키지와 gplot이라는 패키지를 사용할 겁니다.
R의 명령행에 아래 코드를 복사하고 붙여넣기를 하면 자동으로 'limma' 패키지가 설치되고 불러들여 집니다.source("http://bioconductor.org/biocLite.R") biocLite("limma") library(limma)
R의 상단 메뉴 => 패키지들 => 패키지(들) 선택하기 => CRAN mirror: Korea(Seoul) 선택 => Packages: 'gplots' 선택 후 'OK'를 클릭하면 자동으로 'gplots' 패키지가 설치됩니다. 설치가 완료된 후 명령행에서 아래 코드를 입력하면 'gplots' 패키지가 불러들여 집니다.
library(gplots)
자 이제 데이터를 불러와 봅시다. R을 설치하고 실행하면 이런 화면이 나타날 겁니다.
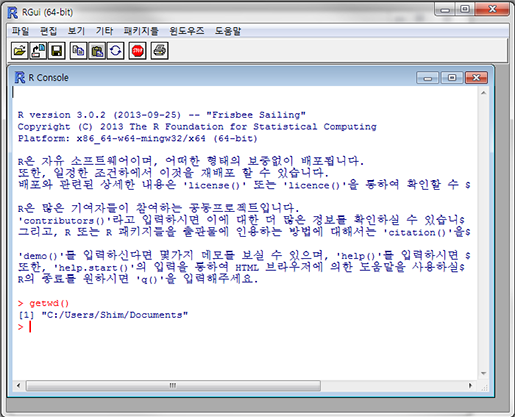
여기에 "getwd()"라고 입력하고 엔터를 치면 이렇게 나타날 겁니다.
> gwtwd() [1] "C:/User/Username/Documents"
이 명령어는 'get working directory', 즉 현재 작업 폴더가 어딘지 출력하라는 명령어입니다. 아까 다운로드한 예제 데이터는 아마도 '바탕화면'이나 '다운로드' 폴더에 있겠죠? 간편하게 실습해 보기 위해 바탕화면에 VennDiagram이라는 새 폴더를 만들고 거기에 예제데이터를 복사해 둡니다. 그리고 'setwd()'라는 명령어로 새로 만든 폴더로 R의 작업 공간을 이동시켜 보겠습니다. 'setwd()'는 아래와 같이 사용하면 됩니다.
> setwd('이동할 폴더 경로')
'이동할 폴더 경로'는 아래와 같은 방법으로 얻을 수 있습니다. 새로 만든 폴더 상단에 주소 입력줄을 클릭해 보면 숨어 있던 상세한 폴더 경로가 나타날 겁니다. 폴더 경로가 전체 선택된 상태에서 'Ctrl+c' 를 눌러 복사를 하고 다시 R로 돌아온 다음 'Ctrl+v'를 눌러 붙여 넣기를 한 키보드의 'Home' 버튼을 눌러 커서를 제일 처음으로 옮겨 명령줄을 아래와 같이 고치고 엔터를 칩니다.
폴더 경로에서 'Users' 다음의 'Username'은 사용자 마다 다릅니다. 붙여 넣기한 그대로 사용하셔야 합니다. 여기서 고쳐져야 하는 것은 역슬레쉬('\') 기호를 하나가 아니라 두개로 입력되어야 하는 점입니다. 윈도우에서는 몇 가지 이유로 R이 역슬레쉬를 그대로 인식하지 못해 이런 방법으로 알려줘야 합니다.
setwd('C:\\Users\\Username\\Desktop\\VennDiagram')
역슬레쉬를 바꿔주지 않으면 이런 에러가 나타날 겁니다.
> setwd('C:\Users\Username\Desktop\VennDiagram')) 에러: "'C:\U"로 시작하는 문자열들 가운데 16진수가 아닌 '\U'가 사용되었습니다
R로 데이터 불러 오기
테이블 형태로 된 데이터를 불러오는 명령어는 'read.table()' 입니다. 먼저 'rfn' 이라는 변수에 파일 이름을 저장합니다. 그리고 'read.table()'을 이용해 데이터를 불러와 'data'라는 변수에 저장하라는 명령어를 사용합니다. 이 때 'read.table()'의 괄호 안에 몇가지 파라메터를 입력해 줍니다.
첫 번째 rfn은 'read.table'이 불러올 파일이름을 나타냅니다. rfn 대신 따옴표 안에 파일이름을 넣어 사용할 수도 있지만 파일 이름은 다른 변수에 저장하여 그 변수명을 바꾸는게 명령형 길이도 줄어들고 여러모로 편리하기 때문에 이런식으로 사용합니다. 두번째 'sep="\t"'는 테이블의 각 셀이 탭문자('\t')로 구분되어 있다고 알려줍니다. 세번째 'header=T'는 첫번째 줄에 column 이름이 있다고 알려줍니다. 여기서는 예제 데이터의 종명이 들어갈 겁니다. 네번째 'row.names=1'은 첫번째 열에 각 데이터의 ID가 있다고 알려주는 파라메터입니다.
테이블 형태의(그리고 탭으로 분리된) 텍스트 파일은 이 명령어를 이용해 R로 불러올 수 있습니다.데이터가 잘 들어 왔는지 확인해 보기 위해서 명령행에 데이터가 저장된 변수명, 'data'를 입력하고 엔터를 치면 전체 데이터를 볼 수 있습니다. 하지만 만약 데이터가 몹시 클 경우 화면이 가득 차거나 불러오다가 컴퓨터가 멈출 수 있으므로 가급적이면 'head()'와 'tail()' 명령어를 이용해 전체 데이터의 앞과 뒤 6줄씩 출력시켜 확인하는 방법을 권장합니다.> rfn <- "ortholog_example.txt" > data <- read.table(rfn, sep='\t', header=T, row.names=1) > data Species.A Species.B Species.C Species.D Clu8 BO016496 BH047N18.14 Clu9 BO014834 RS013419 AT18397457 Clu3 BO014772 RS018960 AT15242221 BH042D24.27 Clu12 BO009902 Clu1 BO008537 AT15242224 BR143G12.16 Clu5 BO008364 RS038406 BH059G16.7 Clu4 BO008360 RS000136 BH006K11.24 Clu20 BO006059 RS040196 AT79324947 Clu13 BO005849 AT15234929 BH064P04.2 Clu7 BO005339 RS002816 Clu19 RS001238 AT15221666 BH015M19.22 Clu6 RS007302 Clu2 BH059C07.32 Clu18 RS008453 AT18420250 Clu17 AT18412659 BH065N19.8 Clu16 RS002645 AT186503429 Clu15 AT18416334 BH001M22.33 Clu14 RS005812 AT145358042 BH050H15.17 Clu11 AT15219226 Clu10 RS004647 BH027F23_27
> head(data) Species.A Species.B Species.C Species.D Clu8 BO016496 BH047N18.14 Clu9 BO014834 RS013419 AT18397457 Clu3 BO014772 RS018960 AT15242221 BH042D24.27 Clu12 BO009902 Clu1 BO008537 AT15242224 BR143G12.16 Clu5 BO008364 RS038406 BH059G16.7 > tail(data) Species.A Species.B Species.C Species.D Clu17 AT18412659 BH065N19.8 Clu16 RS002645 AT186503429 Clu15 AT18416334 BH001M22.33 Clu14 RS005812 AT145358042 BH050H15.17 Clu11 AT15219226 Clu10 RS004647 BH027F23_27
데이터 형식 변형 시키기
벤다이어그램을 그리는데 사용할 명령어는 'vennCounts()'와 'venn()' 입니다. 이 명령어들은 'logical value'로 이루어진 'data.frame' 형식의 데이터를 입력 받습니다. 'logical value'는 'True' 또는 'FALSE', 즉 '참이다', '거짓이다', 또는 '있다', '없다'를 표현하는 값이란 의미입니다.
우리가 불러온 데이터는 그냥 요소(factor)들로 구성되어 있습니다. 이 데이터에서 값이 있으면 참, 없으면 거짓으로 바꿔 ld라는 변수에 저장시킵니다.
이 부분이 이해가 안 되시면 일단 아래 코드만 쭉~ 따라해 보시면 됩니다.> ld <- (data != "") > ld Species.A Species.B Species.C Species.D Clu8 TRUE FALSE FALSE TRUE Clu9 TRUE TRUE TRUE FALSE Clu3 TRUE TRUE TRUE TRUE Clu12 TRUE FALSE FALSE FALSE Clu1 TRUE FALSE TRUE TRUE Clu5 TRUE TRUE FALSE TRUE Clu4 TRUE TRUE FALSE TRUE Clu20 TRUE TRUE TRUE FALSE Clu13 TRUE FALSE TRUE TRUE Clu7 TRUE TRUE FALSE FALSE Clu19 FALSE TRUE TRUE TRUE Clu6 FALSE TRUE FALSE FALSE Clu2 FALSE FALSE FALSE TRUE Clu18 FALSE TRUE TRUE FALSE Clu17 FALSE FALSE TRUE TRUE Clu16 FALSE TRUE TRUE FALSE Clu15 FALSE FALSE TRUE TRUE Clu14 FALSE TRUE TRUE TRUE Clu11 FALSE FALSE TRUE FALSE Clu10 FALSE TRUE FALSE TRUE
이렇게 변환된 데이터 'ld' 를 'as.data.frame' 명령어를 이용해 'data.frame' 이라는 형식으로 변환시켜 'ld.df' 라는 변수에 저장합니다.
> ld.df <- as.data.frame(ld) > ld.df Species.A Species.B Species.C Species.D Clu8 TRUE FALSE FALSE TRUE Clu9 TRUE TRUE TRUE FALSE Clu3 TRUE TRUE TRUE TRUE Clu12 TRUE FALSE FALSE FALSE Clu1 TRUE FALSE TRUE TRUE Clu5 TRUE TRUE FALSE TRUE Clu4 TRUE TRUE FALSE TRUE Clu20 TRUE TRUE TRUE FALSE Clu13 TRUE FALSE TRUE TRUE Clu7 TRUE TRUE FALSE FALSE Clu19 FALSE TRUE TRUE TRUE Clu6 FALSE TRUE FALSE FALSE Clu2 FALSE FALSE FALSE TRUE Clu18 FALSE TRUE TRUE FALSE Clu17 FALSE FALSE TRUE TRUE Clu16 FALSE TRUE TRUE FALSE Clu15 FALSE FALSE TRUE TRUE Clu14 FALSE TRUE TRUE TRUE Clu11 FALSE FALSE TRUE FALSE Clu10 FALSE TRUE FALSE TRUE
특별히 바뀐게 없어 보이지만, 'class()' 명령어를 이용해 data.frame 형식으로 바꾸기 전후를 비교해 보면 무엇이 바뀐지 알 수 있습니다 'data'를 'logical value'로 변환 시키면 그 결과는 'matrix'라는 형식으로 출력됩니다. 이를 'as.data.frame()'이라는 명령어로 'data.frame' 형식으로 바꿔 'ld.df'라는 변수에 저장을 시킨 겁니다.
> class(ld) [1] "matrix" > class(ld.df) [1] "data.frame"
각 집합 및 교집합들의 유전자 수 알아내기
예제 데이터를 보면 어떤 유전자들은 특정 종에서만 나타나는 유전자이고, 어떤 유전자는 다른 종에서도 ortholog 관계의 유전자가 있다고 합니다. 즉 크게 보면 4개의 집합이 있고, 4개의 집합으로 만들어질 수 있는 부분집합(교집합들과 각 집합의 유니크한 원소만 나타내는 부분집합) 16개가 있습니다. 이 부분집합들에 해당하는 유전자 수를 테이블 형태로 확인하는 명령어가 'vennCounts()' 입니다. 이 명령어에 'ld.df' 를 입력하면 아래와 같은 결과가 출력됩니다.
첫번째 줄을 보면 모든 집합(종에서도)에서 없는('0') 원소의 수가 가장 오른쪽 'Counts' 열에 0개 있다고 출력되어 있습니다. 두번째 줄은 다른 종에서는 없고('0') 'Species.D'에서만 있는('1') 원소의 수가 1개 있다고 합니다. 15번째 줄은 'Species.A', 'Species.B', 'Species.C'에서 공통적으로 ortholog 관계의 유전자가 2개 있다고 출력되어 있습니다.> vennCounts(ld.df) Species.A Species.B Species.C Species.D Counts [1,] 0 0 0 0 0 [2,] 0 0 0 1 1 [3,] 0 0 1 0 1 [4,] 0 0 1 1 2 [5,] 0 1 0 0 1 [6,] 0 1 0 1 1 [7,] 0 1 1 0 2 [8,] 0 1 1 1 2 [9,] 1 0 0 0 1 [10,] 1 0 0 1 1 [11,] 1 0 1 0 0 [12,] 1 0 1 1 2 [13,] 1 1 0 0 1 [14,] 1 1 0 1 2 [15,] 1 1 1 0 2 [16,] 1 1 1 1 1 attr(,"class") [1] "VennCounts"
이제 진짜 벤다이어그램을 그려보자
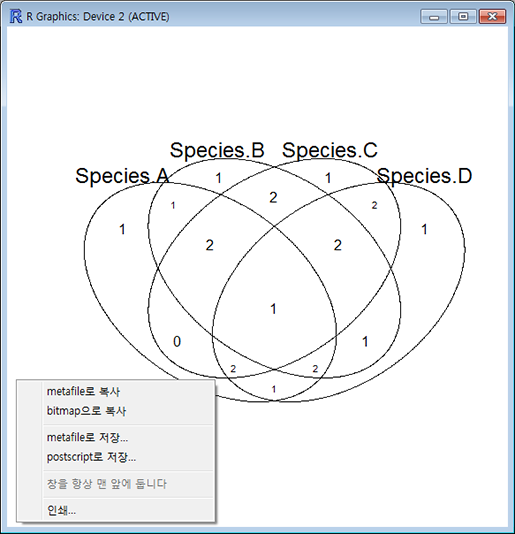
이렇게 준비된 데이터를 벤다이어그램으로 그려주는 명령어는 'venn()' 입니다. 아래 명령을 실행해 보면 자동으로 카운팅된 교집합에 해당하는 원소들의 수가 알맞는 칸에 배치된 벤다이어그램이 출력됩니다. 이렇게 출력된 그림은 창 크기를 적당히 조절해서 사이즈를 조절한 다음 마우스 오른쪽 버튼으로 클릭하면 그림파일로 저장하거나 복사해서 파워포인트로 붙여 넣어 이용할 수 있습니다.
'venn()'은 최대 5개 집합의 데이터만 받아 벤다이어그램을 그려줍니다.> venn(ld.df)
마무리
더 찾아보면 R에서 벤다이어그램을 그려주는 패키지는 상당히 많습니다.
bvenn
http://cran.r-project.org/web/packages/bvenn/index.htmlcolorfulVennPlot
http://cran.r-project.org/web/packages/colorfulVennPlot/index.htmleVenn
http://cran.r-project.org/web/packages/eVenn/index.htmlVennDiagram
http://cran.r-project.org/web/packages/VennDiagram/VennDiagram.pdfVenneuler
http://cran.r-project.org/web/packages/venneuler/index.htmlVennerable
http://goo.gl/YjQCc
이중에서 데이터 조작이 적고, 비교적 간편하게 이용할 수 있는 'gplot'의 'venn()'을 소개해 드렸습니다. 조금더 간편하고 예쁜 벤다이어그램을 그릴 수 있는 방법을 알고 계신 분이 계시면 아래 이메일로 알려주시면 감사하겠습니다. 알려주신 내용으로 업데이트 해서 올리도록 하겠습니다.
가능하면 R이나 코딩에 친숙하지 않은 분들을 위해 작은 부분까지 설명해 드리고자 노력했습니다만, 충분히 전달되지 못한 부분들도 있을 것 같습니다. 이해가 잘 안 되거나, 에러 나는 부분이 있으면 언제든 codes@insilicogen.com 메일 주소로 문의주세요.
여러분의 연구에 작은 도움이 된다면 더할 나위 없이 기쁘겠습니다.Reference
작성자 : Codes사업부 Research팀
심재영 주임
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/141
Trackback URL : 이 글에는 트랙백을 보낼 수 없습니다

