
[Quipu Issue Paper] Assembly Ⅱ - De novo assembly
- Posted at 2010/02/12 08:54
- Filed under 생물정보
연재 순서
1. Assembly
2. Variation study
3. Expression study
4. Epigenomics
5. Genome Annotation
6. Next Generation Bioinformatics
7. Data Management for web 2.0 Era
8. Semantic Network for Integrated Biology Data
9. Gene Network Discovery by Text-mining
10. Centralization for High-throughput Data Analysis
Quipu Issue Paper 기술 소식지 두 번째 연재로 NGS Assembly 중에 De novo assenbly에 대해 알아보도록 하겟습니다.
Human genome project 이후 다양한 종에서 Whole Genome Sequencing(WGS)이 진행되고 있다. 고전적인 방법으로 BAC library를 제작하여 샷건 시퀀싱으로 진행되던 방식이 NGS 시대에 들어 새롭게 진화하였다. 일예로 Dr. Andreas는 ‘Corynebacterium kroppenstedtii’의 유전체 시퀀싱을 단 7.5 시간 만에 수행하고 자동화된 genome annotation 파이프라인을 통해 단 3일 만에 논문으로 발표하였다[13]. 그러나 아쉽게도 미생물을 제외한 대부분의 종에서는 아직까지 NGS를 이용한 de novo assembly로 유전체 시퀀싱을 완성한 팀은 없다. 짧은 reads의 제한적인 정보로 복잡한 유전체 구조를 모두 밝히기엔 어려움이 따른다. 따라서 reference가 없는 새로운 종을 시퀀싱 할 경우에는 짧은 reads를 생성하는 Solexa나 SOLiD보다는 Roche 454를 이용한 long reads 시퀀싱이 유용하다. 2009년 10월 현재 Roche 454의 GS Titanium의 경우 평균 read 길이가 350bp에 달하고 최대 700bp까지 시퀀싱을 수행한다고 한다[8]. 단, 유전체 구조상 반복 서열 영역과 같은 서열상의 정보로만 분석 되지 않는 부분은 paired-end reads의 fragment size를 다양하게 디자인하여 long reads와 함께 분석 하여야 한다. 이렇게 de novo assembly의 경우 long reads와 short paired-end reads를 동시에 처리할 수 있어야 하므로 assembler 또한 이들 모두를 처리할 수 있어야 한다.
대표적인 de novo assembler로 Velvet(Solexa bundle program)[9], Newbler(454 bundle program)[10], ABySS[11], CLC NGS Cell, 그리고 고전적인 프로그램인 Phrap을 들 수 있다. 이들 assembler의 특징에 대해 좀 더 자세히 살펴보기 위해 다음의 몇 가지 조건을 기준으로 살펴보았다. 단, phrap의 경우 NGS reads의 특성상 대량의 데이터를 처리하기엔 메모리와 속도 면에서 비교하기가 어려울 만큼 효율적이지 않은 점을 고려하여 이후 비교 분석에서는 제외하였다.
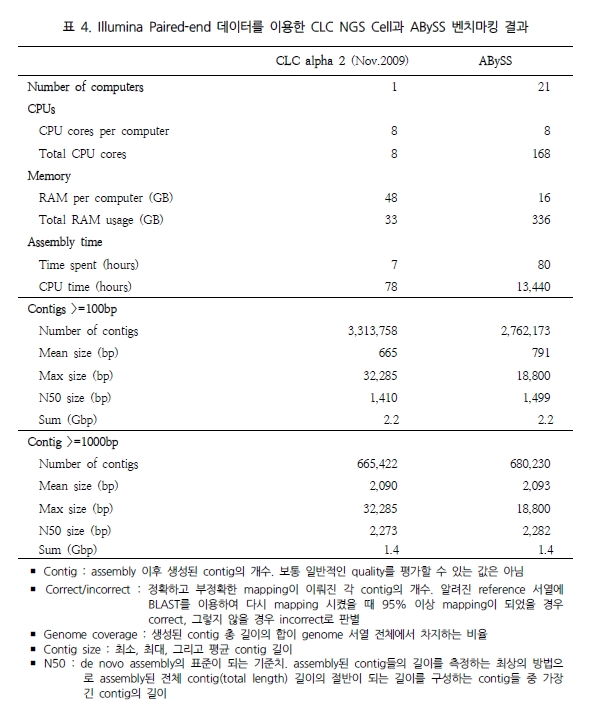
 최근 de novo assembler의 개발이 가속화 되면서 human 유전체를 대상으로 de novo assembly에 성공한 사례가 발표 되었다. CLC NGS Cell[12]과 ABySS[11]가 그 주인공으로 Illumina의 paired-end reads를 분석에 이용하여 38X의 human 유전체를 완성 하였다고 밝혔다. 그 두 프로그램의 결과를 비교해 보면 표 4와 같다. CLC NGS Cell은 최근 2.0에서 3.0 beta 버전으로 업그레이드되면서 de novo assembly에 놀라울 만큼의 결과를 향상 시켰다[12]. 단적으로 38X나 되는 많은 데이터를 de novo assembly로 분석하는데 단 78시간(CPU time)밖에 소요되지 않았다는 것만으로도 매우 놀라운 일이다(표 4).
최근 de novo assembler의 개발이 가속화 되면서 human 유전체를 대상으로 de novo assembly에 성공한 사례가 발표 되었다. CLC NGS Cell[12]과 ABySS[11]가 그 주인공으로 Illumina의 paired-end reads를 분석에 이용하여 38X의 human 유전체를 완성 하였다고 밝혔다. 그 두 프로그램의 결과를 비교해 보면 표 4와 같다. CLC NGS Cell은 최근 2.0에서 3.0 beta 버전으로 업그레이드되면서 de novo assembly에 놀라울 만큼의 결과를 향상 시켰다[12]. 단적으로 38X나 되는 많은 데이터를 de novo assembly로 분석하는데 단 78시간(CPU time)밖에 소요되지 않았다는 것만으로도 매우 놀라운 일이다(표 4).
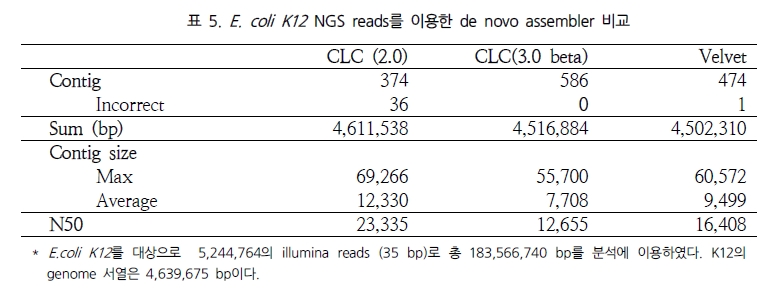
이는 ABySS와 비교했을 때 약 172배가 빨라진 결과이다[12]. 뿐만 아니라 분석된 contig의 품질을 살펴보면 100bp 이상 되는 contig는 ABySS 보다 많으며 최대 contig 길이 면에서 1.7배 긴 contig를 생성하고 있다. N50 또한 서로 비슷한 결과를 보여 주고 있어 단순히 빠른 속도만을 내세우는 프로그램이 아닌 분석 결과에 대한 정확성 면에서도 믿음을 주고 있다. 이를 한 번 더 검증하기 위해 짧은 유전체를 대상으로 Velvet과의 정확성 테스트를 다시 수행하였다. 그 결과 Velvet의 부정확한 assembly에 비해 CLC NGS Cell은 모두 정확한 assembly를 수행하였음을 확인 할 수 있었다(표 5)[12].
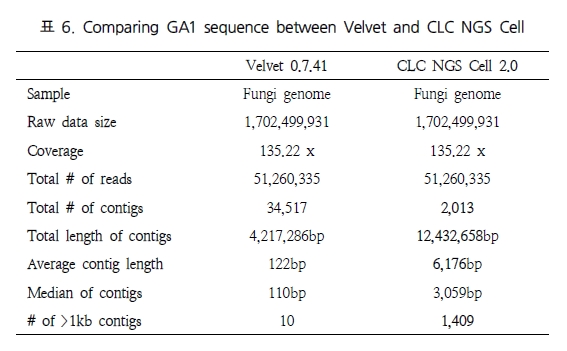
비슷한 결과로 Shizosaccharomyces pombe 132, Fungi 유전체를 대상으로 테스트한 결과에서도 CLC NGS Cell이 Velvet 보다는 좋은 결과를 보였다(표 6). 마지막으로 long reads와 short reads를 동시에 분석하여 복잡한 유전체 구조를 분석 할 때 서로 다른 데이터 플랫폼이 함께 분석되어야 한다. 이를 위해 GS titanium과 Illumina 데이터(Solexa)를 다양한 비율로 구성한 테스트 세트를 이용하여 분석하였다(표 7).
 분석 결과 long reads 구성이 많을수록 긴 contig를 구성하는 것을 확인할 수 있었다. 그러나 여기서 보여지진 않았으나 반복서열 영역과 같은 시퀀싱이 쉽지 않은 영역의 데이터를 long reads 보다는 short reads에서 확인할 수 있었다. 따라서 두 가지 플랫폼의 장점을 모두 수용할 수 있는 assembler를 선택하여 분석의 정확성을 높이는 것이 좋을 듯하다.
분석 결과 long reads 구성이 많을수록 긴 contig를 구성하는 것을 확인할 수 있었다. 그러나 여기서 보여지진 않았으나 반복서열 영역과 같은 시퀀싱이 쉽지 않은 영역의 데이터를 long reads 보다는 short reads에서 확인할 수 있었다. 따라서 두 가지 플랫폼의 장점을 모두 수용할 수 있는 assembler를 선택하여 분석의 정확성을 높이는 것이 좋을 듯하다.
NGS 데이터의 분석 단계는 크게 pre-processing, assembly, 그리고 assembly를 이용한 이차 분석으로 나눠진다. Pre-processing 단계에서는 다양한 플랫폼으로부터 single reads, long reads, paired reads 그리고 unpaired reads들의 정보를 assembly 단계에 적용하기 위한 작업을 수행한다. 대부분의 assembler는 대용량의 데이터 처리를 위해 index 파일을 자체 프로그램에 맞게 생산하는 단계를 거치거나, 다양한 플랫폼에서 생산된 데이터를 특정 포맷의 입력 포맷으로 전환하는 과정을 수행한다. 그러나 이러한 과정은 자칫 시퀀싱 자체의 raw 정보를 유실하는 경우가 발생할 수 있으므로 assembler의 기능을 면밀히 살펴 최대한 정보를 그대로 보존할 수 있는 assembler를 선택하는 것이 좋다. 그중 CLC NGS Cell은 대부분의 시퀀싱 raw 파일을 입력 포맷으로 지원하므로 이러한 정보 손실을 줄여 줄 수 있는 이점이 있다. 더욱이 zip file 형태의 파일을 바로 입력 포맷으로 지원하므로 분석 단계에서의 파일 관리가 수월한 점도 장점이라 하겠다.
다음으로 assembly 과정에 대해 알아보자. NGS reads의 assembly는 제한적인 computing power를 고려하여 데이터를 여러 개로 분리하여 반복 수행하게 된다. 이후 이들 assembly 결과를 하나로 합치는 과정을 통해 전체적인 assembly을 완성한다. 대부분의 프로그램이 한 번의 명령어 수행으로 contig 서열 혹은 assembly 파일을 얻을 수 있다. 그림 3. CLC NGS Cell workflow. 다양한 입력 포맷을 지원하므로 assembly 수행을 위한 여러 단계의 전처리 과정이 없으며 assembly 이후 한 번의 스크립트 수행을 통해 원하는 다양한 정보를 이차적으로 생산할 수 있다.
마지막으로 assembly 결과를 이용한 다양한 이차정보 분석이다. SNP와 같은 variation 분석, assembly 결과를 보여주는 그래픽 인터페이스 그리고 assembly quality 정보 분석이 주로 수행된다. 그 중 assembly quality는 reference assembly의 경우 assembly에 참여된 reads의 coverage와 fold로 나타낼 수 있으며 de novo assembly의 경우 N50 및 fold value가 지표가 될 수 있다. 이러한 분석 역시 간단한 명령어 수행으로 대부분의 프로그램에서 수행하고 있다(그림 3).
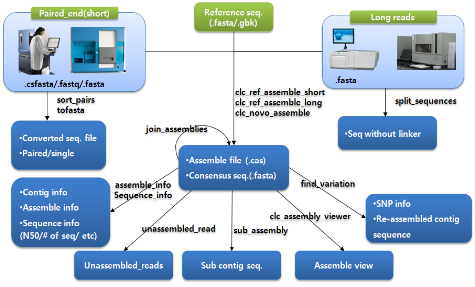 그림 3. CLC NGS Cell workflow. 다양한 입력 포맷을 지원하므로 assembly 수행을 위한 여러 단계의 전처리 과정이 없으며 assembly 이후 한 번의 스크립트 수행을 통해 원하는 다양한 정보를 이차적으로 생산할 수 있다.
그림 3. CLC NGS Cell workflow. 다양한 입력 포맷을 지원하므로 assembly 수행을 위한 여러 단계의 전처리 과정이 없으며 assembly 이후 한 번의 스크립트 수행을 통해 원하는 다양한 정보를 이차적으로 생산할 수 있다.
또한 그림 4는 alignment 결과와 그에 따른 SNP evidence를 그래픽 인터페이스를 통해 보여주고 있다. CLC NGS Cell은 reference assembly 수행 시 유전자 구조 및 기능 정보를 담고 있는 NCBI의 genbank 포맷의 파일을 reference 파일로 입력 받을 수 있는데, 이를 이용하게 되면 assembly 수행 후 결과를 CLC Genomics Workbench를 통해 유전자 위치와 alignment 된 reads 정보를 따로 그래픽 인터페이스를 제작하지 않고도 쉽게 확인 할 수 있다. 또한 SNP 정보를 함께 CLC Genomics Workbench를 통해 확인할 수 있어 바로 프라이머를 제작하는 등의 차후 분석이 가능하도록 돕고 있다.
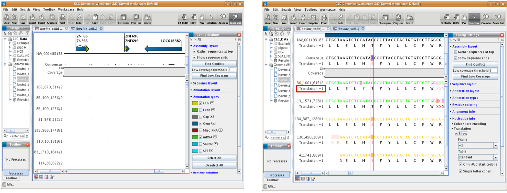 그림 4. CLC Genomics Workbench를 이용한 alignment view 와 SNP view. Reference assembly 수행 시 annotation 정보가 있는 .gbk 파일을 이용하여 분석한 후 assembly 파일을 Genomics Workbench를 통해 확인하면 유전자의 위치와 함께 alignment reads의 상세정보를 확인 할 수 있다. 아울러 SNP 정보 중 cSNP의 경우 translation 정보를 활용하여
그림 4. CLC Genomics Workbench를 이용한 alignment view 와 SNP view. Reference assembly 수행 시 annotation 정보가 있는 .gbk 파일을 이용하여 분석한 후 assembly 파일을 Genomics Workbench를 통해 확인하면 유전자의 위치와 함께 alignment reads의 상세정보를 확인 할 수 있다. 아울러 SNP 정보 중 cSNP의 경우 translation 정보를 활용하여
non-synonymous/synonymous SNP를 구분하여 분석 할 수 있다.
다음주 연재에서는 Assembly에 이어서 Assembly를 수행하고 이후 서열간의 비교 분석을 통해 variation 분석을 진행하는 variation study에 대해 알아보도록 하겠습니다.
많은 관심 부탁드립니다.
참고문헌
1. Li R, Li Y, Kristiansen K, Wang J. (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714 (http://soap.genomics.org.cn/index.html)
2. Li H, Ruan J, Durbin R. (2008) Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18, 1851–1858 (http://maq.sourceforge.net/index.shtml)
3. Lin H, Zhang Z, Zhang MQ, Ma B, Li M. (2008) ZOOM! Zillions of oligos mapped. Bioinformatics 24, 2431–2437 (http://www.bioinfor.com)
4. CLC NGS Cell : http://www.clcbio.com
5. White paper on reference assembly on the CLC NGS Cell 2.0 (www.clcbio.com)
6. Palmieri N, Schlötterer C. (2009) Mapping accuracy of short reads from massively parallel sequencing and the implications for quantitative expression profiling. PLoS One. 28, 4(7):e6323.
7. Ning Z, Cox AJ, Mullikin JC. (2001) SSAHA: a fast search method for large DNA databases. Genome Res. 10, 1725-1729.
8. Roche 454 : http://www.454.com/
9. Zerbino DR, Birney E. (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829.(http://www.ebi.ac.uk/~zerbino/velvet/)
10. Newbler : 454 bundle program
11. Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, Stazyk G, Morin RD, Zhao Y, Hirst M, Schein JE, Horsman DE, Connors JM, Gascoyne RD, Marra MA, Jones SJ. (2009) De novo transcriptome assembly with ABySS. Bioinformatics. 21, 2872-2877
12. White paper on de novo assembly in CLC NGS Cell 3.0 beta (www.clcbio.com)
13. Andreas T., Eva T., Thomas B., Alexander G., Ulrike L. and Alfred P. Ultrafast de novo sequencing of the human pathogen Corynebacterium urealyticum with the Genome Sequencer System (http://www.454.com/downloads/protocols/Whole_Genome_Sequencing_And_Assembly.pdf)
14. Horner DS, Pavesi G, Castrignanò T, De Meo PD, Liuni S, Sammeth M, Picardi E, Pesole G. (2009) Bioinformatics approaches for genomics and post genomics applications of next-generation sequencing. Brief Bioinform. [Epub ahead of print]
15. Kent WJ. (2002) BLAT--the BLAST-like alignment tool. Genome Res. 4, 656-664.
16. Ning Z, Cox AJ, Mullikin JC. (2001) SSAHA: a fast search method for large DNA databases. Genome Res. 10, 1725-1729.
17. Langmead B, Trapnell C, Pop M, Salzburg SL. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 3, R25
18. Jiang H, Wong WH. (2008) SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 20, 395-396.
1. Assembly
2. Variation study
3. Expression study
4. Epigenomics
5. Genome Annotation
6. Next Generation Bioinformatics
7. Data Management for web 2.0 Era
8. Semantic Network for Integrated Biology Data
9. Gene Network Discovery by Text-mining
10. Centralization for High-throughput Data Analysis
Quipu Issue Paper 기술 소식지 두 번째 연재로 NGS Assembly 중에 De novo assenbly에 대해 알아보도록 하겟습니다.
1-1-2. De novo assembly
Human genome project 이후 다양한 종에서 Whole Genome Sequencing(WGS)이 진행되고 있다. 고전적인 방법으로 BAC library를 제작하여 샷건 시퀀싱으로 진행되던 방식이 NGS 시대에 들어 새롭게 진화하였다. 일예로 Dr. Andreas는 ‘Corynebacterium kroppenstedtii’의 유전체 시퀀싱을 단 7.5 시간 만에 수행하고 자동화된 genome annotation 파이프라인을 통해 단 3일 만에 논문으로 발표하였다[13]. 그러나 아쉽게도 미생물을 제외한 대부분의 종에서는 아직까지 NGS를 이용한 de novo assembly로 유전체 시퀀싱을 완성한 팀은 없다. 짧은 reads의 제한적인 정보로 복잡한 유전체 구조를 모두 밝히기엔 어려움이 따른다. 따라서 reference가 없는 새로운 종을 시퀀싱 할 경우에는 짧은 reads를 생성하는 Solexa나 SOLiD보다는 Roche 454를 이용한 long reads 시퀀싱이 유용하다. 2009년 10월 현재 Roche 454의 GS Titanium의 경우 평균 read 길이가 350bp에 달하고 최대 700bp까지 시퀀싱을 수행한다고 한다[8]. 단, 유전체 구조상 반복 서열 영역과 같은 서열상의 정보로만 분석 되지 않는 부분은 paired-end reads의 fragment size를 다양하게 디자인하여 long reads와 함께 분석 하여야 한다. 이렇게 de novo assembly의 경우 long reads와 short paired-end reads를 동시에 처리할 수 있어야 하므로 assembler 또한 이들 모두를 처리할 수 있어야 한다.
대표적인 de novo assembler로 Velvet(Solexa bundle program)[9], Newbler(454 bundle program)[10], ABySS[11], CLC NGS Cell, 그리고 고전적인 프로그램인 Phrap을 들 수 있다. 이들 assembler의 특징에 대해 좀 더 자세히 살펴보기 위해 다음의 몇 가지 조건을 기준으로 살펴보았다. 단, phrap의 경우 NGS reads의 특성상 대량의 데이터를 처리하기엔 메모리와 속도 면에서 비교하기가 어려울 만큼 효율적이지 않은 점을 고려하여 이후 비교 분석에서는 제외하였다.
이는 ABySS와 비교했을 때 약 172배가 빨라진 결과이다[12]. 뿐만 아니라 분석된 contig의 품질을 살펴보면 100bp 이상 되는 contig는 ABySS 보다 많으며 최대 contig 길이 면에서 1.7배 긴 contig를 생성하고 있다. N50 또한 서로 비슷한 결과를 보여 주고 있어 단순히 빠른 속도만을 내세우는 프로그램이 아닌 분석 결과에 대한 정확성 면에서도 믿음을 주고 있다. 이를 한 번 더 검증하기 위해 짧은 유전체를 대상으로 Velvet과의 정확성 테스트를 다시 수행하였다. 그 결과 Velvet의 부정확한 assembly에 비해 CLC NGS Cell은 모두 정확한 assembly를 수행하였음을 확인 할 수 있었다(표 5)[12].
비슷한 결과로 Shizosaccharomyces pombe 132, Fungi 유전체를 대상으로 테스트한 결과에서도 CLC NGS Cell이 Velvet 보다는 좋은 결과를 보였다(표 6). 마지막으로 long reads와 short reads를 동시에 분석하여 복잡한 유전체 구조를 분석 할 때 서로 다른 데이터 플랫폼이 함께 분석되어야 한다. 이를 위해 GS titanium과 Illumina 데이터(Solexa)를 다양한 비율로 구성한 테스트 세트를 이용하여 분석하였다(표 7).
1-1-3. Workflow
NGS 데이터의 분석 단계는 크게 pre-processing, assembly, 그리고 assembly를 이용한 이차 분석으로 나눠진다. Pre-processing 단계에서는 다양한 플랫폼으로부터 single reads, long reads, paired reads 그리고 unpaired reads들의 정보를 assembly 단계에 적용하기 위한 작업을 수행한다. 대부분의 assembler는 대용량의 데이터 처리를 위해 index 파일을 자체 프로그램에 맞게 생산하는 단계를 거치거나, 다양한 플랫폼에서 생산된 데이터를 특정 포맷의 입력 포맷으로 전환하는 과정을 수행한다. 그러나 이러한 과정은 자칫 시퀀싱 자체의 raw 정보를 유실하는 경우가 발생할 수 있으므로 assembler의 기능을 면밀히 살펴 최대한 정보를 그대로 보존할 수 있는 assembler를 선택하는 것이 좋다. 그중 CLC NGS Cell은 대부분의 시퀀싱 raw 파일을 입력 포맷으로 지원하므로 이러한 정보 손실을 줄여 줄 수 있는 이점이 있다. 더욱이 zip file 형태의 파일을 바로 입력 포맷으로 지원하므로 분석 단계에서의 파일 관리가 수월한 점도 장점이라 하겠다.
다음으로 assembly 과정에 대해 알아보자. NGS reads의 assembly는 제한적인 computing power를 고려하여 데이터를 여러 개로 분리하여 반복 수행하게 된다. 이후 이들 assembly 결과를 하나로 합치는 과정을 통해 전체적인 assembly을 완성한다. 대부분의 프로그램이 한 번의 명령어 수행으로 contig 서열 혹은 assembly 파일을 얻을 수 있다. 그림 3. CLC NGS Cell workflow. 다양한 입력 포맷을 지원하므로 assembly 수행을 위한 여러 단계의 전처리 과정이 없으며 assembly 이후 한 번의 스크립트 수행을 통해 원하는 다양한 정보를 이차적으로 생산할 수 있다.
마지막으로 assembly 결과를 이용한 다양한 이차정보 분석이다. SNP와 같은 variation 분석, assembly 결과를 보여주는 그래픽 인터페이스 그리고 assembly quality 정보 분석이 주로 수행된다. 그 중 assembly quality는 reference assembly의 경우 assembly에 참여된 reads의 coverage와 fold로 나타낼 수 있으며 de novo assembly의 경우 N50 및 fold value가 지표가 될 수 있다. 이러한 분석 역시 간단한 명령어 수행으로 대부분의 프로그램에서 수행하고 있다(그림 3).
또한 그림 4는 alignment 결과와 그에 따른 SNP evidence를 그래픽 인터페이스를 통해 보여주고 있다. CLC NGS Cell은 reference assembly 수행 시 유전자 구조 및 기능 정보를 담고 있는 NCBI의 genbank 포맷의 파일을 reference 파일로 입력 받을 수 있는데, 이를 이용하게 되면 assembly 수행 후 결과를 CLC Genomics Workbench를 통해 유전자 위치와 alignment 된 reads 정보를 따로 그래픽 인터페이스를 제작하지 않고도 쉽게 확인 할 수 있다. 또한 SNP 정보를 함께 CLC Genomics Workbench를 통해 확인할 수 있어 바로 프라이머를 제작하는 등의 차후 분석이 가능하도록 돕고 있다.
non-synonymous/synonymous SNP를 구분하여 분석 할 수 있다.
다음주 연재에서는 Assembly에 이어서 Assembly를 수행하고 이후 서열간의 비교 분석을 통해 variation 분석을 진행하는 variation study에 대해 알아보도록 하겠습니다.
많은 관심 부탁드립니다.
참고문헌
1. Li R, Li Y, Kristiansen K, Wang J. (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714 (http://soap.genomics.org.cn/index.html)
2. Li H, Ruan J, Durbin R. (2008) Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18, 1851–1858 (http://maq.sourceforge.net/index.shtml)
3. Lin H, Zhang Z, Zhang MQ, Ma B, Li M. (2008) ZOOM! Zillions of oligos mapped. Bioinformatics 24, 2431–2437 (http://www.bioinfor.com)
4. CLC NGS Cell : http://www.clcbio.com
5. White paper on reference assembly on the CLC NGS Cell 2.0 (www.clcbio.com)
6. Palmieri N, Schlötterer C. (2009) Mapping accuracy of short reads from massively parallel sequencing and the implications for quantitative expression profiling. PLoS One. 28, 4(7):e6323.
7. Ning Z, Cox AJ, Mullikin JC. (2001) SSAHA: a fast search method for large DNA databases. Genome Res. 10, 1725-1729.
8. Roche 454 : http://www.454.com/
9. Zerbino DR, Birney E. (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829.(http://www.ebi.ac.uk/~zerbino/velvet/)
10. Newbler : 454 bundle program
11. Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, Stazyk G, Morin RD, Zhao Y, Hirst M, Schein JE, Horsman DE, Connors JM, Gascoyne RD, Marra MA, Jones SJ. (2009) De novo transcriptome assembly with ABySS. Bioinformatics. 21, 2872-2877
12. White paper on de novo assembly in CLC NGS Cell 3.0 beta (www.clcbio.com)
13. Andreas T., Eva T., Thomas B., Alexander G., Ulrike L. and Alfred P. Ultrafast de novo sequencing of the human pathogen Corynebacterium urealyticum with the Genome Sequencer System (http://www.454.com/downloads/protocols/Whole_Genome_Sequencing_And_Assembly.pdf)
14. Horner DS, Pavesi G, Castrignanò T, De Meo PD, Liuni S, Sammeth M, Picardi E, Pesole G. (2009) Bioinformatics approaches for genomics and post genomics applications of next-generation sequencing. Brief Bioinform. [Epub ahead of print]
15. Kent WJ. (2002) BLAT--the BLAST-like alignment tool. Genome Res. 4, 656-664.
16. Ning Z, Cox AJ, Mullikin JC. (2001) SSAHA: a fast search method for large DNA databases. Genome Res. 10, 1725-1729.
17. Langmead B, Trapnell C, Pop M, Salzburg SL. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 3, R25
18. Jiang H, Wong WH. (2008) SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 20, 395-396.
Posted by 人Co
- Tag
- 454, Assembler, de novo assembly, NGS, Solexa, WGS, 생물정보학, 인실리코젠
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/37
Trackback URL : 이 글에는 트랙백을 보낼 수 없습니다

