
나만 알고 싶은 OmicsBox
- Posted at 2021/04/30 17:17
- Filed under 제품소식

'유전체 데이터는 점차 쌓여가고···쌓여가는 데이터 처리는 해야겠고···이왕 처리하는 거 효율적으로, 내가 원하는 방향으로 처리하고 싶은데···심도 있는 분석은 또 어떻게 진행해야 하는 걸까···' 이런 의문 품어보신 적 다들 있으시죠?! 있을 겁니다! 저 또한 그랬으니까요!
제 경우에는 OmicsBox라는 솔루션이 저의 이러한 의문을 해소해주었습니다. 제가 OmicsBox를 처음 접한 건 바야흐로 대학원 시절. 그 당시에는 OmicsBox로 리뉴얼 되기 전인 Blast2GO 버전이었습니다. 저는 그 당시 De Novo Transcriptome 분석을 진행했었고, BLAST 분석 이후의 후속 분석인 Functional Analysis(GO, KEGG, InterProScan)를 Blast2GO를 이용하여 진행했었습니다.
만약 그 당시의 저에게 Blast2GO가 없었다면, 저는 아마 졸업을 못 했을 겁니다. (생각만 해도 끔찍하네요.)
서론이 길었네요 자, 그럼 이렇게 저를 무사히 졸업시켜주는데 일조했던 OmicsBox(구 Blast2GO)! 과연 어떤 기능을 하는 생물정보 솔루션인지 이제부터 차근차근 알아볼까요?
자, 그럼 이렇게 저를 무사히 졸업시켜주는데 일조했던 OmicsBox(구 Blast2GO)! 과연 어떤 기능을 하는 생물정보 솔루션인지 이제부터 차근차근 알아볼까요?
나만 알고 싶은 OmicsBox
OmicsBox 기능을 알아보기 전에 OmicsBox는 무엇인지에 대해 먼저 알아보겠습니다.
OmicsBox는 새로운 genome 분석을 위한 최고의 생물정보학 플랫폼(Platform)임과 동시에 산업, 학술 및 정부 연구기관의 생물학자를 위한 사용자 친화적인 생물정보학 데스크톱 애플리케이션(Desktop application)입니다. 이러한 OmicsBox는 기능 유전체학의 선두주자로서 세계적으로 인정받고 있으며, 이는 7,000개 이상의 과학 연구 인용으로 입증되었습니다. 또한, OmicsBox는 genomics, transcriptomics, metagenomics의 NGS 데이터 분석에 최적화(de novo 파트)되어 있으며, 필요에 따라 데이터 분석에 필요한 다양한 모듈(Module)을 결합하여 사용할 수 있습니다.
제 경우에는 OmicsBox라는 솔루션이 저의 이러한 의문을 해소해주었습니다. 제가 OmicsBox를 처음 접한 건 바야흐로 대학원 시절. 그 당시에는 OmicsBox로 리뉴얼 되기 전인 Blast2GO 버전이었습니다. 저는 그 당시 De Novo Transcriptome 분석을 진행했었고, BLAST 분석 이후의 후속 분석인 Functional Analysis(GO, KEGG, InterProScan)를 Blast2GO를 이용하여 진행했었습니다.
만약 그 당시의 저에게 Blast2GO가 없었다면, 저는 아마 졸업을 못 했을 겁니다. (생각만 해도 끔찍하네요.)
서론이 길었네요
나만 알고 싶은 OmicsBox
OmicsBox 기능을 알아보기 전에 OmicsBox는 무엇인지에 대해 먼저 알아보겠습니다.
OmicsBox는 새로운 genome 분석을 위한 최고의 생물정보학 플랫폼(Platform)임과 동시에 산업, 학술 및 정부 연구기관의 생물학자를 위한 사용자 친화적인 생물정보학 데스크톱 애플리케이션(Desktop application)입니다. 이러한 OmicsBox는 기능 유전체학의 선두주자로서 세계적으로 인정받고 있으며, 이는 7,000개 이상의 과학 연구 인용으로 입증되었습니다. 또한, OmicsBox는 genomics, transcriptomics, metagenomics의 NGS 데이터 분석에 최적화(de novo 파트)되어 있으며, 필요에 따라 데이터 분석에 필요한 다양한 모듈(Module)을 결합하여 사용할 수 있습니다.
[그림 1]. OmicsBox OverView
- 아래는 OmicsBox의 주요 기능을 나열해 봤는데요,
- 클라우드 플랫폼(Cloud platform)을 활용한 애플리케이션(Application) 고속 실행 및 견고하고 안전한 백엔드(Back-end) 제공
- Blast 결과의 Gene Ontology mapping
- Functional Annotation
- InterProScan domain 검색
- GO-Slim Reduction
- KEGG map mapping
- 통계 정보 차트
- 다양한 데이터의 import와 export format 지원
- Eukaryote, prokaryote에 알맞은 model을 이용한 유전자 부위 예측
- 발현값을 이용한 pairwise/time course 별 분석
이뿐만 아니라 OmicsBox는 아래와 같은 강력한 장점도 가지고 있습니다.
Advantages of OmicsBox
Powerful Tables
Workflows
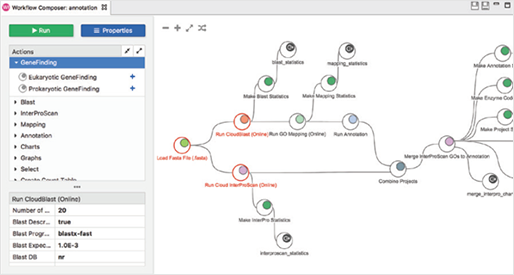
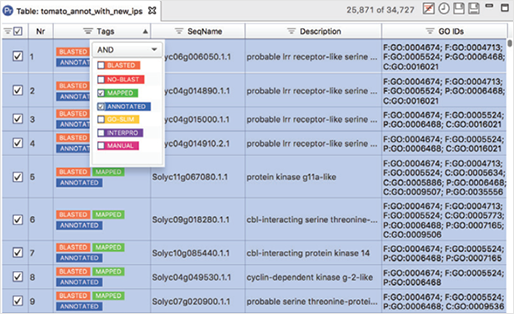
Workflow manager를 사용하여 생물정보학적 workflow를 생성, 실행 및 저장할 수 있으며, 선택한 분석 단계를 끌어와 workflow를 생성하고 모든 파라미터(Parameter)를 한 곳에서 구성하고 검토할 수 있습니다.
Genome Browser
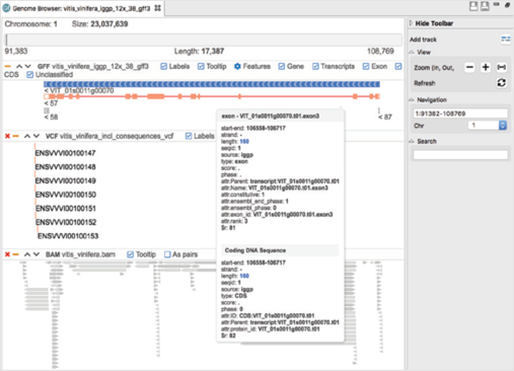
Genome Browser는 여러 트랙(Track)을 통해 alignment(.bam), gene annotation(.gff) 및 variant 정보(.vcf)를 결합할 수 있습니다. 그뿐만 아니라 navigation, 필터 및 검색 옵션을 사용하여 쉽고 탐색적인 방법으로 결과를 확인할 수 있습니다.
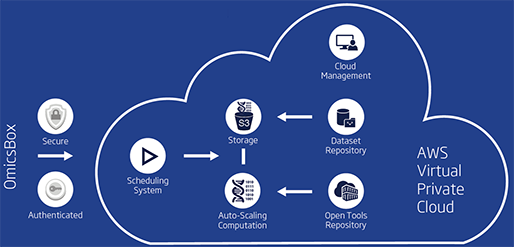
Omics Cloud Platform
Advantages of OmicsBox
Powerful Tables
[그림 2]. Powerful Tables
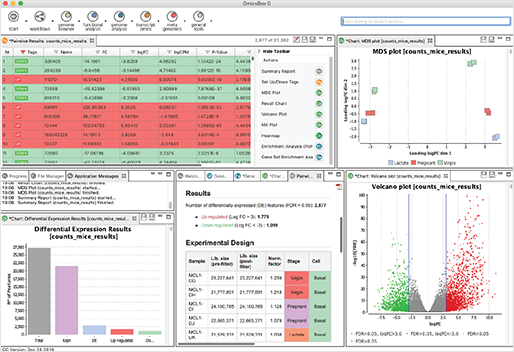
풍부한 사용자 인터페이스(Interface)를 통해 대용량 데이터세트를 쉽게 처리할 수 있고, 모든 테이블은 필터링과 정렬을 할 수 있으며, 가장 중요한 것은 다른 결과 세트와 결합할 수 있다는 것입니다. 또한, 후속 분석 단계의 하위 집합을 쉽게 생성하고 추출할 수 있습니다.
Workflows
[그림 3]. Workflows
Workflow manager를 사용하여 생물정보학적 workflow를 생성, 실행 및 저장할 수 있으며, 선택한 분석 단계를 끌어와 workflow를 생성하고 모든 파라미터(Parameter)를 한 곳에서 구성하고 검토할 수 있습니다.
[그림 4]. Genome Browser
Genome Browser는 여러 트랙(Track)을 통해 alignment(.bam), gene annotation(.gff) 및 variant 정보(.vcf)를 결합할 수 있습니다. 그뿐만 아니라 navigation, 필터 및 검색 옵션을 사용하여 쉽고 탐색적인 방법으로 결과를 확인할 수 있습니다.
Omics Cloud Platform
[그림 5]. Omics Cloud Platform
클라우드 플랫폼(Cloud platform)은 대부분의 무거운 작업이 수행되는 OmicsBox에 견고하고 안전하며 자동 확장이 가능한 백엔드(Back-end)를 제공합니다. 또한, 이 시스템을 사용하면 표준 PC에서 매우 까다로운 생물정보학 애플리케이션(Application)을 고속으로 실행할 수 있습니다.
자 어떤가요? 전반적으로 OmicsBox에 대해 간략히 알아보았는데, 글을 읽다 보니 내 데이터를 어떻게 처리하면 좋을지 구상이 잡히셨나요?  아직 잘 안 잡히셨다고요? 괜찮습니다.
아직 잘 안 잡히셨다고요? 괜찮습니다.")
이제부터 설명해 드릴 OmicsBox의 4가지 모듈을 살펴보고 나면 구상이 잘 잡히실 겁니다.
이제부터 설명해 드릴 OmicsBox의 4가지 모듈을 살펴보고 나면 구상이 잘 잡히실 겁니다.
그럼 한번 살펴볼까요?
Four modules of OmicsBox
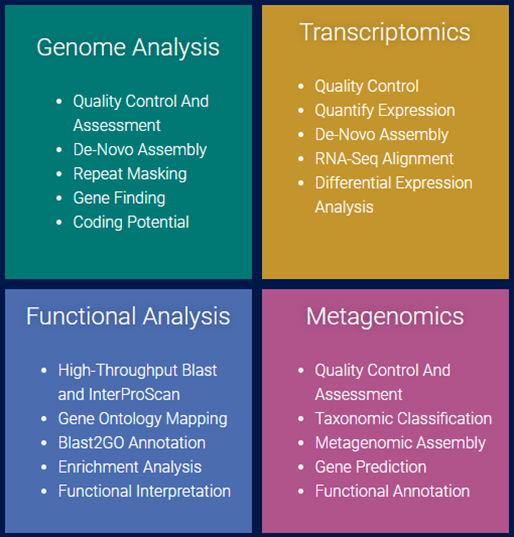
Genome Analysis
[그림 6]. Four modules of OmicsBox
Genome Analysis
- Quality Control : 샘플의 품질 관리를 수행하기 위해, FastQC와 Trimmomatic을 사용하여 Reads를 필터링하고, low quality bases를 제거할 수 있습니다.
- De Novo Assembly : ABySS를 기반으로 하는 assembly 기능을 통해 reference genome 또는 특정 하드웨어(Hardware) 요구 사항 없이 전체 genome sequence를 재구성할 수 있습니다.
- Repeat Masking : 다운스트림(Down-stream) 유전자 예측을 개선하기 위해 RepeatMasker를 사용하여 진핵생물 genome의 반복적이고 복잡성이 낮은 assemble된 DNA sequence를 마스킹(Masking)할 수 있습니다.
- Gene Finding : Genome 구조를 특성화하기 위해 원핵생물(Glimmer 사용) 및 진핵생물(Augustus 사용) 유전자 예측을 수행할 수 있으며, 진핵생물 유전자 예측은 RNA-seq 인트론 힌트를 지원합니다.
- Genome Browser : 트랙(Track) 형태로 annotation을 시각화하여 genome sequences(.fasta), alignments(.bam), intron-exon structure(.gff) 및 variant data(.vcf)와 결합할 수 있습니다.
Transcriptomics
- Quality Control : 샘플의 품질 관리를 수행하기 위해, FastQC와 Trimmomatic을 사용하여 Reads를 필터링하고 low quality bases를 제거할 수 있습니다.
- De Novo Assembly : Reference genome 없이 de novo transcriptome을 생성하기 위해 Trinity 프로그램을 이용하여 짧은 Reads를 조립할 수 있습니다.
- RNA-Seq Alignment : 초고속 유니버설(Universal) RNA-seq aligner인 STAR를 사용하여 RNA-seq 데이터를 reference genome에 alignment 할 수 있습니다.
- Quantify Expression : HTSeq 또는 RSEM을 사용하여 reference genome의 유무와 관계없이 gene 또는 transcript 수준에서의 발현을 정량화할 수 있습니다.
- Differential Expression Analysis : NOISeq, edgeR 또는 maSigPro와 같이 잘 알려진 다양한 통계 패키지(Package)를 사용하여 실험 조건 간 또는 시간이 지남에 따라 차등적으로 발현된 유전자를 검출할 수 있습니다. 또한, 풍부한 시각화는 결과를 해석하는 데 많은 도움이 됩니다.
- Enrichment Analysis : 차등 발현 결과를 functional annotation과 결합함으로써, Enrichment 분석은 과잉 및 과소 표현된 생물학적 기능을 식별할 수 있도록 해줍니다.
Metagenomics
- Quality Control : 샘플의 품질 관리를 수행하기 위해, FastQC와 Trimmomatic을 사용하여 Reads를 필터링하고 low quality bases를 제거할 수 있습니다.
- Taxonomic Classification : Kraken에서 현재의 종(세균, 고세균, 바이러스)을 식별하고, 다단계의 Pie chart인 Krona와 샘플 간의 비교 막대 그래프로 결과를 시각화할 수 있습니다.
- Metagenomics Assembly : 클라우드(Cloud)에서 빠르고 쉽게 대규모 데이터세트를 조립하기 위해 MetaSPAdes와 MEGAHIT 중에서 선택할 수 있습니다.
- Gene Prediction : 가능 유전자와 단백질을 식별하고 추출하기 위해 일반 Reads에는 FragGeneScan, 조립된 데이터에는 Prodigal을 사용할 수 있습니다.
- Functional Interpretation : EggNOG-Mapper 및 PfamScan을 사용하여 높은 처리량의 functional annotation을 얻을 수 있고, 결과를 GO graph 및 chart로 시각적으로 표현하고 비교할 수 있습니다.
Functional Analysis
- High-Throughput Blast and InterProScan : CloudBlast 및 CloudInterProScan을 사용하여 선택한 reference 데이터세트에 대해 빠른 sequence alignment 및 domain 검색을 수행할 수 있습니다.
- Gene Ontology Mapping : UniProt 및 Gene Ontology Consortia의 최신 데이터베이스에서 사용 가능한 functional annotation을 사용하여 잠재적인 homologous와 domain을 연결할 수 있습니다.
- Blast2GO Annotation : Blast2GO 방법론을 사용함으로써, source annotation 품질 및 ontology 계층을 고려하여 가장 신뢰할 수 있는 기능 label을 새로운 sequence 데이터세트에 유연하게 할당할 수 있습니다.
- Enrichment Analysis : 서로 다른 enrichment 분석 방법 (Fisher Exact Test 및 GSEA)을 사용하여 과다 및 과소 표현된 분자 기능을 식별할 수 있습니다.
- Functional Interpretation : 다양한 시각화를 통해 annotation process를 평가할 수 있을 뿐만 아니라 실험 및 기능 분석 결과의 생물학적 해석을 도울 수 있습니다.
Genome 분석, Transciptome 분석, Metagenome 분석 받고 Functional Annotation 분석까지! NGS 분석 대부분 분야를 섭렵한 OmicsBox, 어떠신 것 같나요? 구상이 어느 정도 잡히시고 있나요? 점점 OmicsBox 매력에 빠져들고 계신가요?
그럼 이쯤에서 이런 질문을 던질 수도 있습니다. "Genome 분석, Transcriptome 분석, Metagenome 분석, Functional Annotation 분석까지 기능은 매우 좋은데 어떻게 이용해야 할지 잘 모르겠어요···"
걱정하지 마세요! 저희에게는 앞서 언급되었던 장점 중 하나인 workflows가 있습니다!
OmicsBox Workflows
Genome Analysis Workflows
Genome Analysis Workflows
- Eukaryotic Genome Analysis Workflow
- Prokaryotic Genome Analysis Workflow
- Long Reads Eukaryotic Genome Analysis Workflow
- Long Reads Prokaryotic Genome Analysis Workflow
[그림 7]. Eukaryotic Genome Analysis Workflow
Transcriptomics Workflows
- De Novo Transcriptome Characterization Workflow
- Transcript-level Analysis Workflow
- Gene-level Analysis Workflow

[그림 8]. De Novo Transcriptome Characterizatioin Workflow
Metagenomics Workflows
- Taxonomic Classification Workflow
- Functional Annotationi Workflow
[그림 9]. Taxonomic Classification Workflow
Metagenomics Workflows
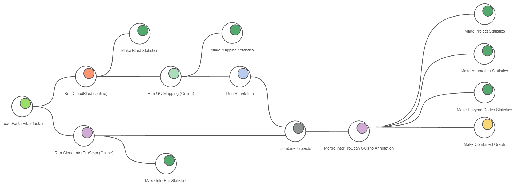
[그림 10]. Functional Analysis Workflow
Raw data만 넣어주고, 각 단계의 파라미터(Parameter)만 잡아주면~ 자동으로 output까지 산출되는 workflow! 참으로 간단하죠?
OmicsBox workflow만 있으면 어렵던 유전체 데이터 분석이 앞으로는 재미있게 느껴질 수 있습니다.
마치며
이번 포스팅(Posting)에서는 OMICS 데이터 분석을 용이하게 해주는 생물정보학 솔루션 OmicsBox에 대해 알아보았는데요. Genome 분석부터 Functional Annotation 분석까지 많은 기능이 있는 아주 매력적인 솔루션이라고 생각이 됩니다. 더군다나 어떤 기능을 어떻게 사용해야 하는지 잘 모르더라도 강력한 기능인 workflow가 있어서 손쉽게 output을 얻을 수도 있구요. 만약 NGS 데이터 분석이 아직 어렵고 낯설게 느껴지는 분들이 계신다면 이번 포스팅의 주인공인 OmicsBox를 강력! 추천해 드립니다. 후회하지 않으실 거에요
만약 OmicsBox와 친해지고 싶으시다면 consulting@insilicogen.com으로 메일 주세요.
친절히 안내 드리도록 하겠습니다.
마지막으로 긴 글 읽어주셔서 감사드리며, 모두 건강한 나날 보내시길 바랍니다.
감사합니다.
OmicsBox : BIOINFORMATICS MADE EASY

만약 OmicsBox와 친해지고 싶으시다면 consulting@insilicogen.com으로 메일 주세요.
친절히 안내 드리도록 하겠습니다.
마지막으로 긴 글 읽어주셔서 감사드리며, 모두 건강한 나날 보내시길 바랍니다.
감사합니다.
OmicsBox : BIOINFORMATICS MADE EASY
[그림 11]. OmicsBox Logo
참고자료
- https://www.biobam.com/omicsbox/
- https://www.biobam.com/wpcontent/uploads/2019/05/OmicsBox_Brochure_BioIT_2019.pdf
- http://manual.omicsbox.biobam.com/user-manual/
- Insilicogen, Inc.
- SCH
작성 : iLAB 조항철 주임컨설턴트
Posted by 人Co
- Tag
- biobam, Bioinformatics, OmicsBox, SCH, 생물정보, 소프트웨어, 인실리코젠
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/380
Trackback URL : 이 글에는 트랙백을 보낼 수 없습니다

